

Review
doi: 10.1172/JCI70212.
Epub 2013 Aug 1.
Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer
Affiliations
- PMID: 23908119
- PMCID: PMC3726176
- DOI: 10.1172/JCI70212
Item in Clipboard
Review
Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer
J Clin Invest.
2013 Aug.
Abstract
Four decades ago, angiogenesis was recognized as a therapeutic target for blocking cancer growth. Because of its importance, VEGF has been at the center stage of antiangiogenic therapy. Now, several years after FDA approval of an anti-VEGF antibody as the first antiangiogenic agent, many patients with cancer and ocular neovascularization have benefited from VEGF-targeted therapy; however, this anticancer strategy is challenged by insufficient efficacy, intrinsic refractoriness, and resistance. Here, we examine recent discoveries of new mechanisms underlying angiogenesis, discuss successes and challenges of current antiangiogenic therapy, and highlight emerging antiangiogenic paradigms.
Figures
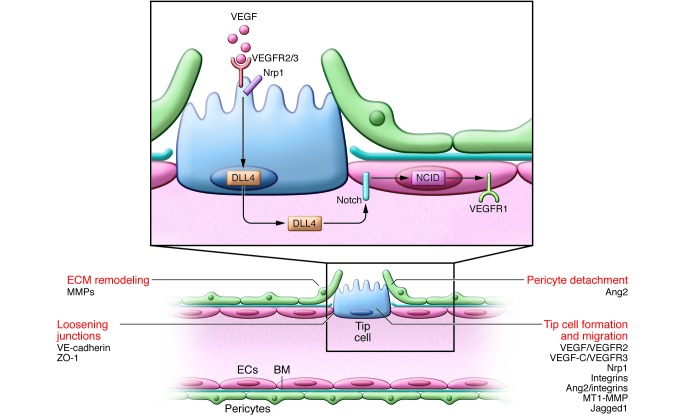
Vascular sprouting is initiated by proangiogenic factors (e.g., VEGF). ECs at the leading edge of the vascular sprout extend filopodia and migrate toward angiogenic signals. VEGF activates VEGFR2 to stimulate tip cell migration. The coreceptor Nrp1 complexes with and enhances VEGFR2 signaling. ECs become either the migratory vessel-leading tip cell or the proliferating stalk cell, but their phenotype is fluid; Notch regulates this specification. ECs with activated VEGFR2 signaling compete for the tip cell position by increasing their expression of DLL4, which binds to Notch receptors on neighboring ECs, releasing the transcription regulator NICD. NICD transcriptionally downregulates VEGFR2 and Nrp1 expression while increasing VEGFR1, a VEGF trap, thus enhancing the stalk cells’ unresponsiveness to VEGF. The tip cell is not a fixed position, and fluidity at the front occurs depending on the VEGFR1/VEGFR2 ratio. Tip cell migration requires BM degradation (in part due to MMP), EC junction loosening (caused by VE-cadherin, ZO-1, and others), and pericyte detachment (regulated by Ang2). VEGF increases the permeability of the vessel, allowing the extravasation of plasma proteins (e.g., fibronectin and fibrinogen) that are deposited as a provisional matrix layer while the preexisting interstitial matrix is remodeled by proteases; these events enable tip cell migration. Key molecular players discussed in this review and elsewhere (5, 132) are indicated.
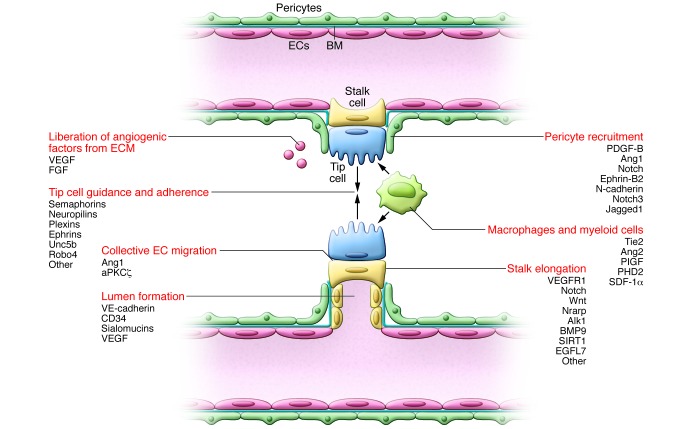
The growing sprout moves along a VEGF gradient. Tip cells adhere to the ECM, mediated by integrins, and migrate toward guidance signal molecules (e.g., semaphorins and ephrins). Stalk cells trail behind the tip cell and proliferate to allow sprout elongation and lumen formation. While Notch signaling inhibits proliferation, expression of Nrarp at branch points allows Wnt signaling to maintain stalk cell proliferation. This system allows vascular migration/directionality (by tip cells) and elongation of the shaft (by proliferating stalk cells). When two tip cells meet, they fuse (anastomose); this mechanism is assisted by macrophages, which accumulate at sites of vascular anastomosis to act as bridge cells by interacting with the neighboring tip cells’ filopodia. Once contact between the tip cells has been established, VE-cadherin–containing junctions further strengthen the connection. Perivascular macrophages further stimulate sprouting by producing angiogenic factors or proteolytically liberating them from the ECM. The stalk cells also deposit BM and recruit pericytes, thus stabilizing the forming vessel. Pericyte precursors are attracted to vessels by EC-expressed PDGF. Once at the vessel, these mesenchymal precursor cells differentiate to pericytes in response to TGF-β and decrease EC migration, proliferation, and vascular leakage, resulting in nascent vessel stabilization. Key molecular players discussed in this Review and elsewhere (5, 132) are indicated.
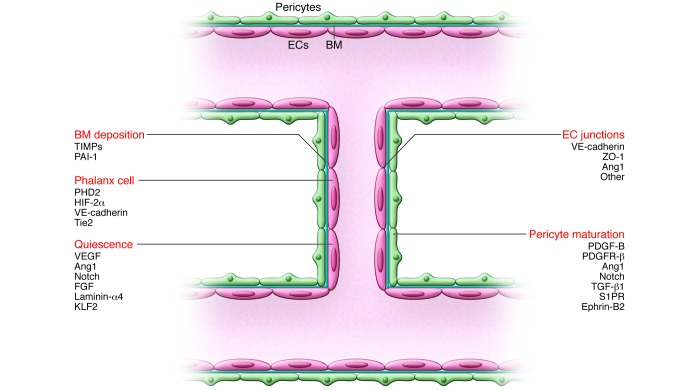
Once fusion has occurred, a connected lumen is formed to allow blood flow through the new vessel. This perfuses the hypoxic tissue, and the resultant oxygen and nutrient delivery leads to decreased levels of angiogenic signals, inactivation of EC oxygen sensors, and increased proquiescent molecules that lead to EC quiescence. Establishment of the blood flow remodels vessel connections, which are regulated by the shear stress–responsive transcription factor KLF2. ECs resume a quiescent phalanx phenotype in a tightly apposed monolayer with a streamlined surface that conducts the blood flow and regulates tissue perfusion. Perfusion induces vascular maturation by reestablishment of cell-cell junctions, pericyte maturation, and BM deposition. Autocrine and paracrine signaling from ECs and surrounding support cells by VEGF, FGF, Ang1, and Notch, among others, maintain a quiescent EC phenotype and protect the vessel from environmental stresses. Reduced growth factor signaling can lead to vessel retraction and EC apoptosis. Once stabilized and matured, the vessel forms a barrier between the blood and surrounding tissue, controlling the exchange of fluids and solutes. Key molecular players discussed in this Review and elsewhere (5, 132) are indicated.
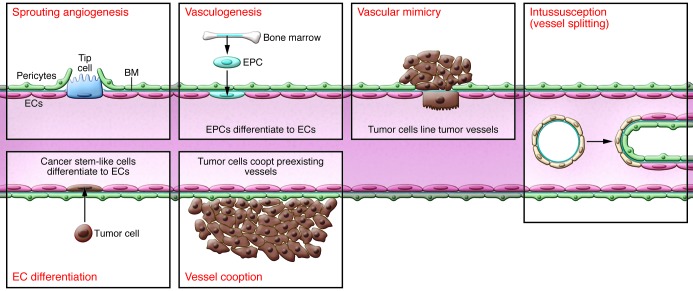
After development, the vasculature rarely extends, but does so in tumor formation. Tumor vascularization occurs via a number of potential mechanisms. While angiogenesis is the most investigated, and the focus of this Review, other mechanisms have been observed. Endothelial progenitor cells (EPCs), which can either reside in the vascular wall or migrate from bone marrow in response to chemoattractants from the tumor cell, can differentiate into ECs and contribute to vessel formation. Vascular mimicry can also occur, whereby tumor cells can act as replacement cells for ECs. Another possibility is that chromosomal abnormalities in putative cancer stems cells allows tumor cells to differentiate into ECs. Other mechanisms by which tumor cells can obtain a blood flow include vessel cooption, whereby the tumor cell arises near to (or migrates toward) a preexisting blood vessel, or the process of intussusception, whereby a preformed vessel splits into two daughter vessels by the insertion of a tissue pillar.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
