

AMPD2 regulates GTP synthesis and is mutated in a potentially treatable neurodegenerative brainstem disorder
- PMID: 23911318
- PMCID: PMC3815927
- DOI: 10.1016/j.cell.2013.07.005
AMPD2 regulates GTP synthesis and is mutated in a potentially treatable neurodegenerative brainstem disorder
Abstract
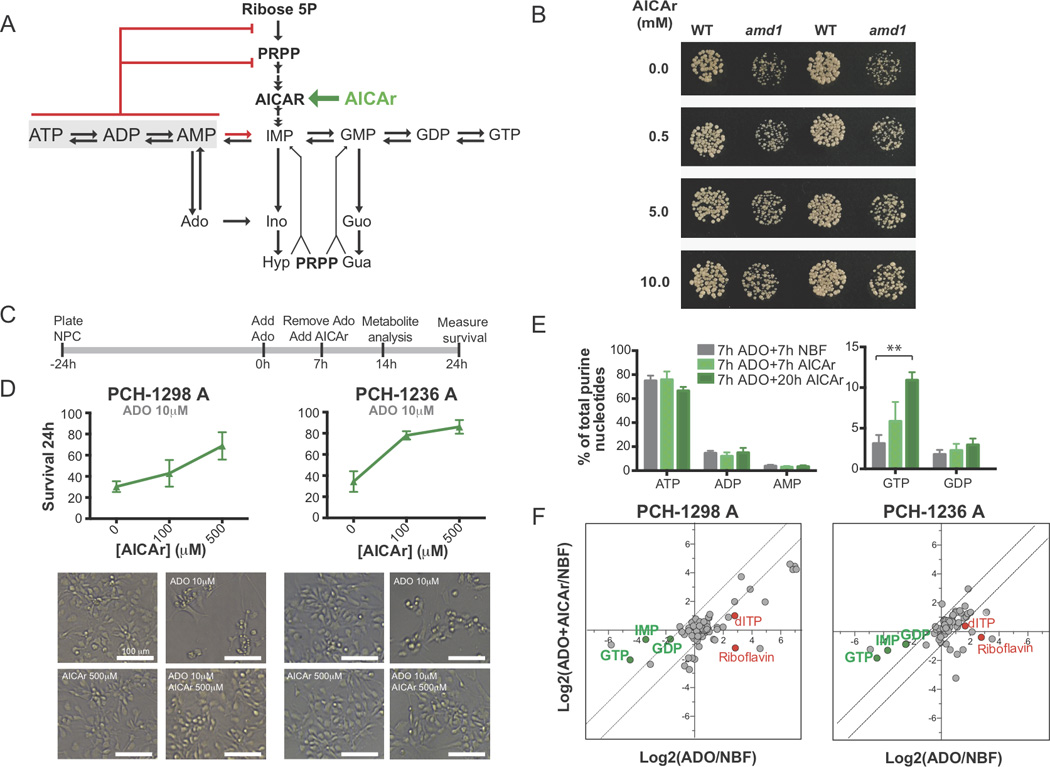
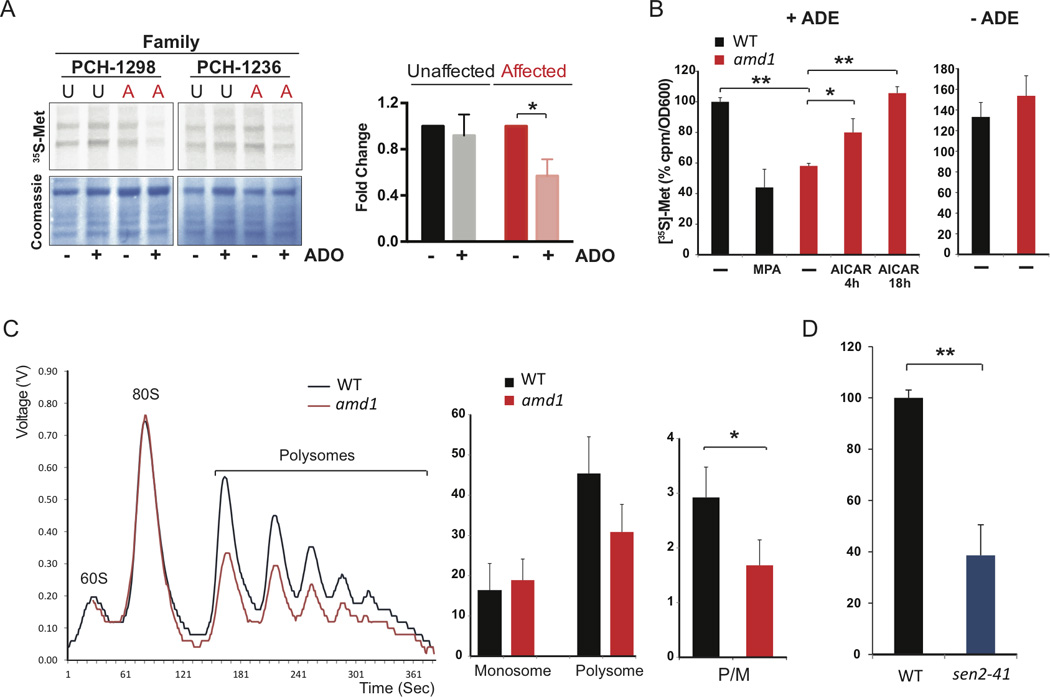
Purine biosynthesis and metabolism, conserved in all living organisms, is essential for cellular energy homeostasis and nucleic acid synthesis. The de novo synthesis of purine precursors is under tight negative feedback regulation mediated by adenosine and guanine nucleotides. We describe a distinct early-onset neurodegenerative condition resulting from mutations in the adenosine monophosphate deaminase 2 gene (AMPD2). Patients have characteristic brain imaging features of pontocerebellar hypoplasia (PCH) due to loss of brainstem and cerebellar parenchyma. We found that AMPD2 plays an evolutionary conserved role in the maintenance of cellular guanine nucleotide pools by regulating the feedback inhibition of adenosine derivatives on de novo purine synthesis. AMPD2 deficiency results in defective GTP-dependent initiation of protein translation, which can be rescued by administration of purine precursors. These data suggest AMPD2-related PCH as a potentially treatable early-onset neurodegenerative disease.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures





References
-
- Alfonzo JD, Sahota A, Taylor MW. Purification and characterization of adenine phosphoribosyltransferase from Saccharomyces cerevisiae. Biochim Biophys Acta. 1997;1341:173–182. - PubMed
-
- Archer S, Juranka PF, Ho JH, Chan VL. An analysis of multiple mechanisms of adenosine toxicity in baby hamster kidney cells. J Cell Physiol. 1985;124:226–232. - PubMed
-
- Barth PG. Pontocerebellar hypoplasias. An overview of a group of inherited neurodegenerative disorders with fetal onset. Brain Dev. 1993;15:411–422. - PubMed
-
- Bausch-Jurken MT, Mahnke-Zizelman DK, Morisaki T, Sabina RL. Molecular cloning of AMP deaminase isoform L. Sequence and bacterial expression of human AMPD2 cDNA. J Biol Chem. 1992;267:22407–22413. - PubMed
-
- Budde BS, Namavar Y, Barth PG, Poll-The BT, Nurnberg G, Becker C, van Ruissen F, Weterman MA, Fluiter K, te Beek ET, et al. tRNA splicing endonuclease mutations cause pontocerebellar hypoplasia. Nat Genet. 2008;40:1113–1118. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
