

BET bromodomains mediate transcriptional pause release in heart failure
- PMID: 23911322
- PMCID: PMC4090947
- DOI: 10.1016/j.cell.2013.07.013
BET bromodomains mediate transcriptional pause release in heart failure
Abstract
Heart failure (HF) is driven by the interplay between regulatory transcription factors and dynamic alterations in chromatin structure. Pathologic gene transactivation in HF is associated with recruitment of histone acetyl-transferases and local chromatin hyperacetylation. We therefore assessed the role of acetyl-lysine reader proteins, or bromodomains, in HF. Using a chemical genetic approach, we establish a central role for BET family bromodomain proteins in gene control during HF pathogenesis. BET inhibition potently suppresses cardiomyocyte hypertrophy in vitro and pathologic cardiac remodeling in vivo. Integrative transcriptional and epigenomic analyses reveal that BET proteins function mechanistically as pause-release factors critical to expression of genes that are central to HF pathogenesis and relevant to the pathobiology of failing human hearts. This study implicates epigenetic readers as essential effectors of transcriptional pause release during HF pathogenesis and identifies BET coactivator proteins as therapeutic targets in the heart.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
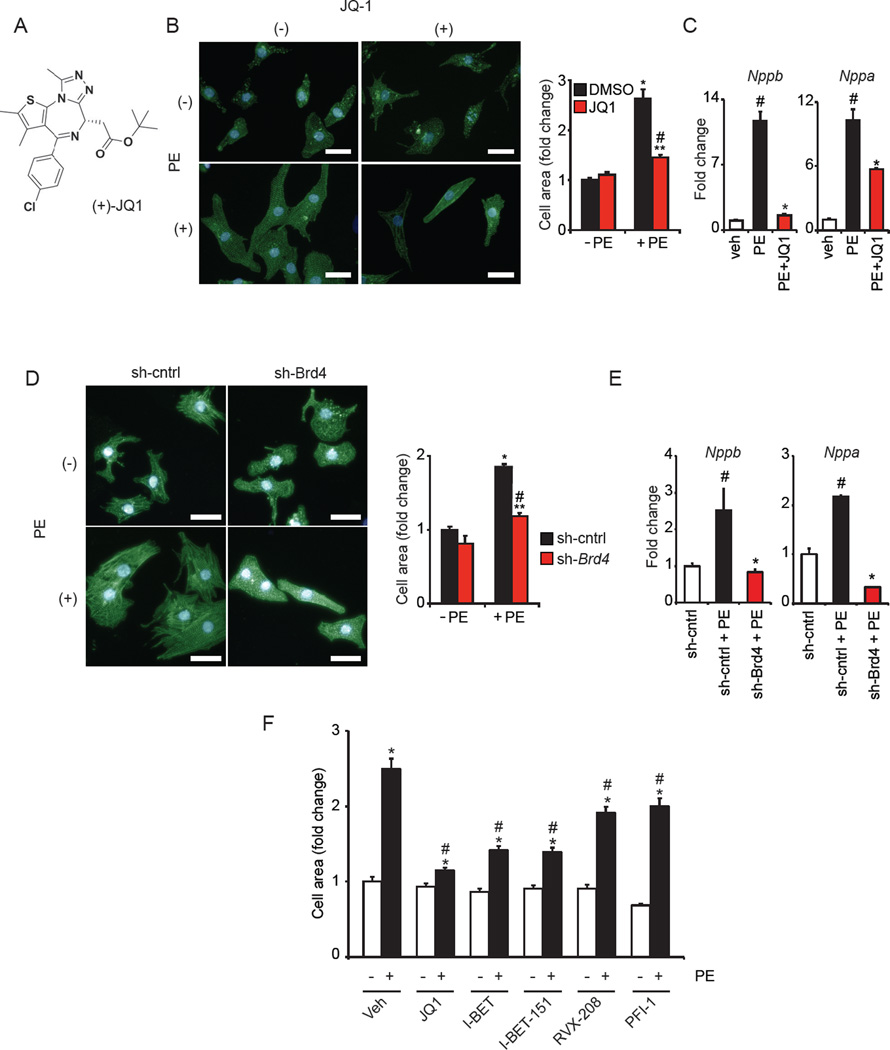
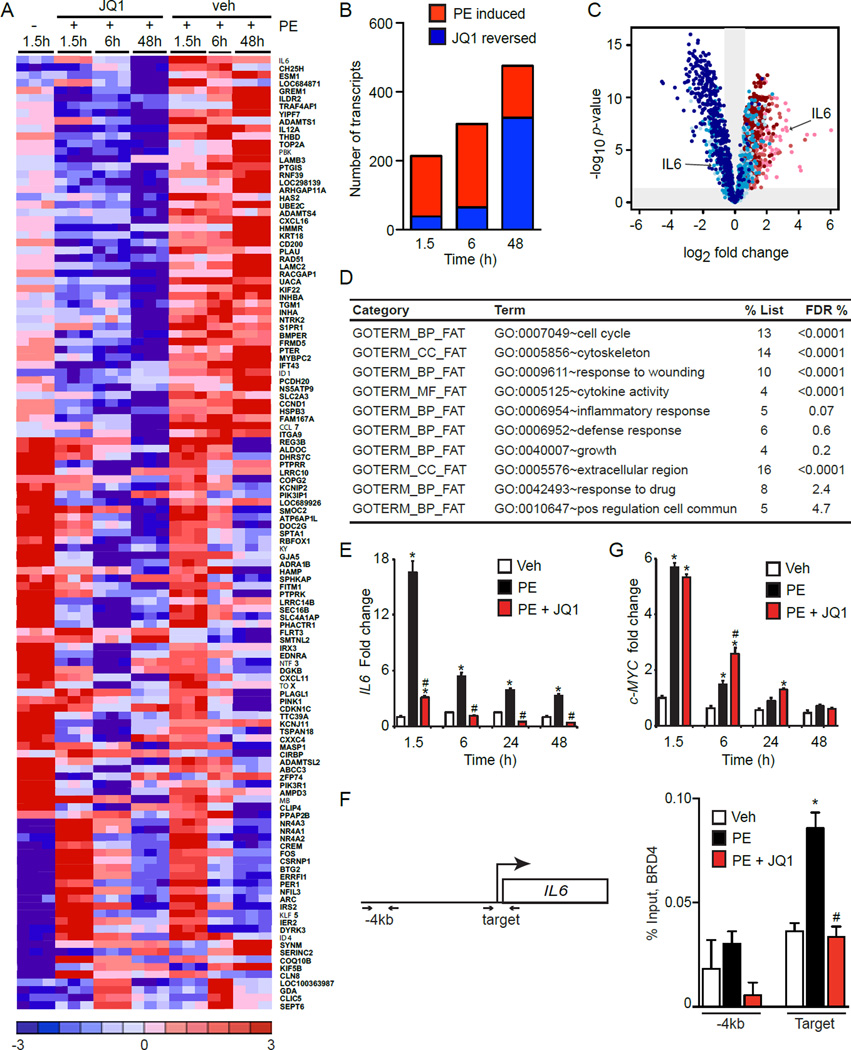
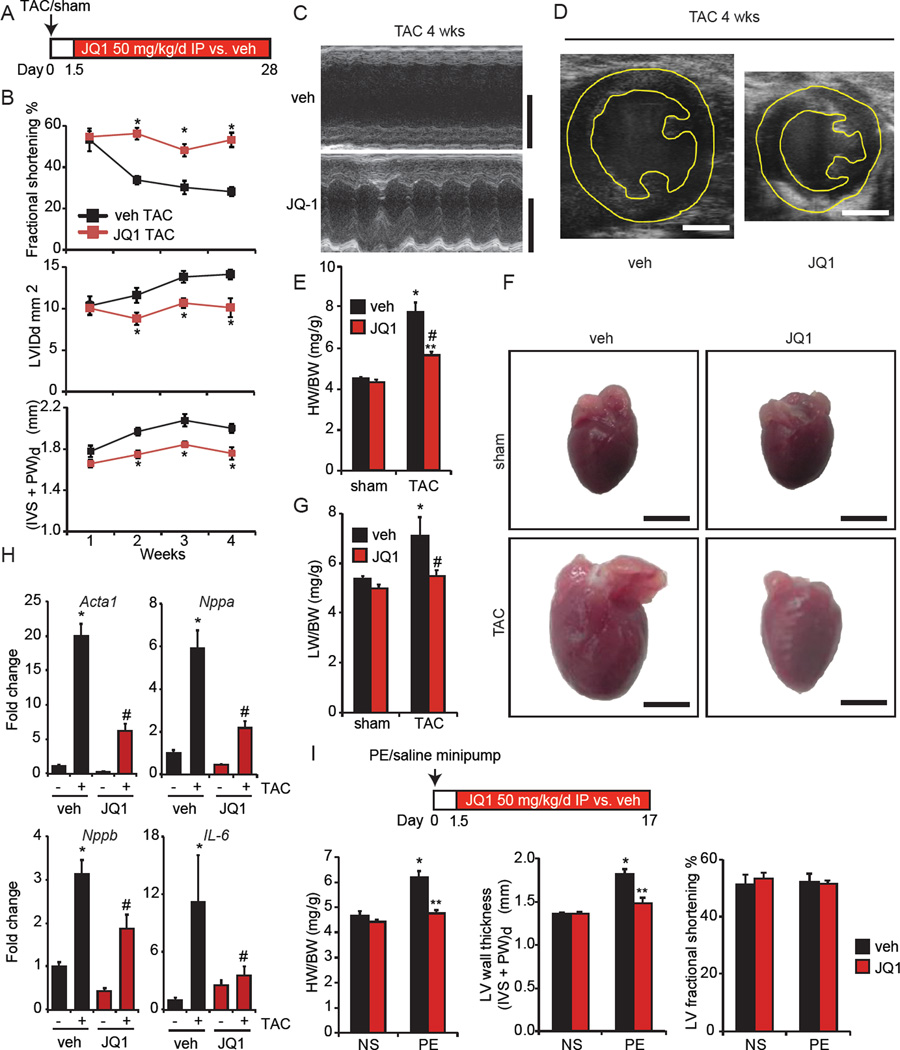
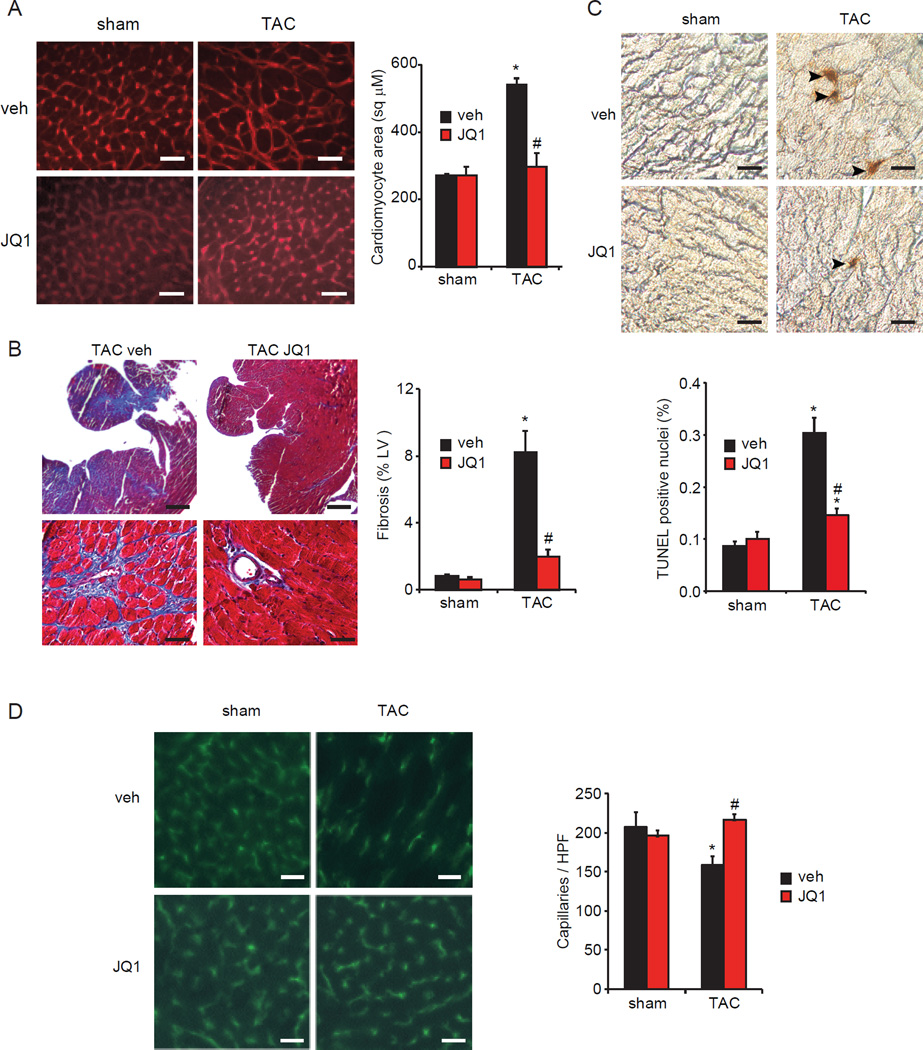
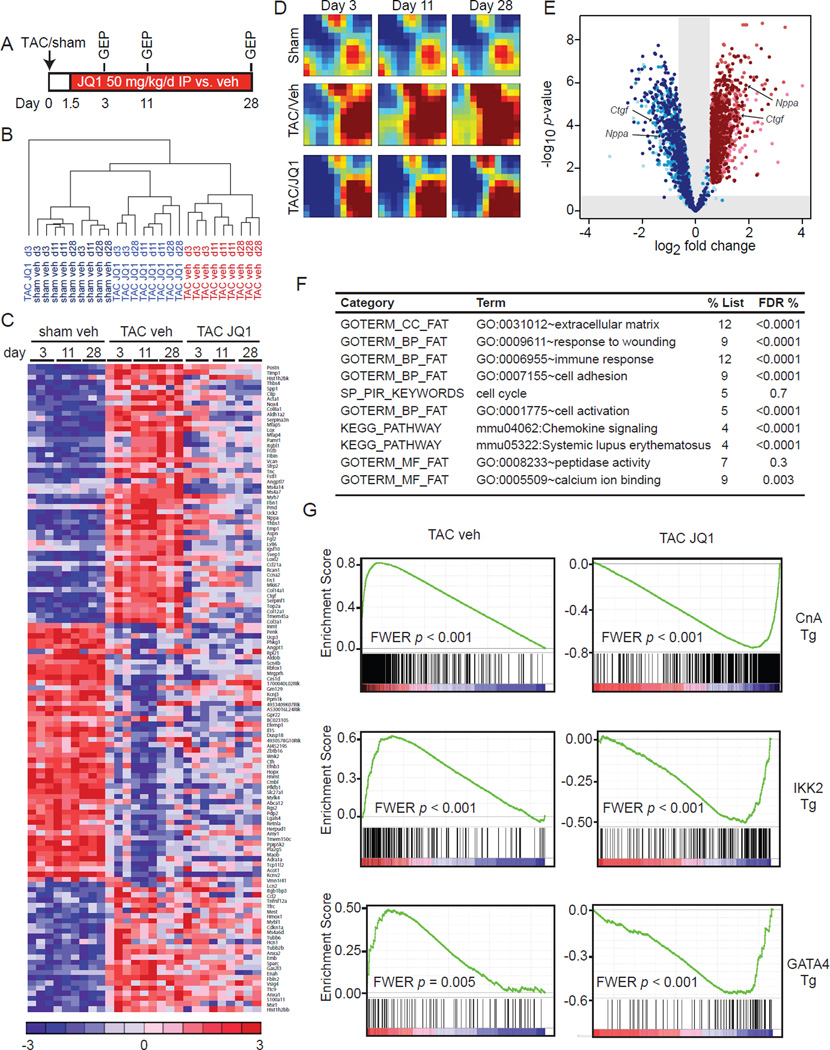
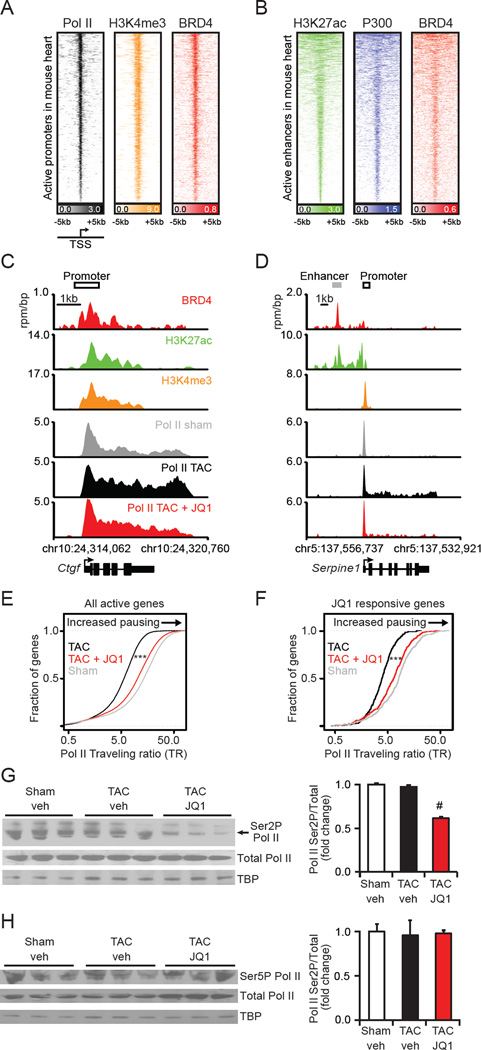
Comment in
-
Cardiology: Bromodomain inhibition halts heart failure.Nat Rev Drug Discov. 2013 Oct;12(10):740. doi: 10.1038/nrd4134. Nat Rev Drug Discov. 2013. PMID: 24080694 No abstract available.
References
-
- Bousette N, Chugh S, Fong V, Isserlin R, Kim KH, Volchuk A, Backx PH, Liu P, Kislinger T, MacLennan DH, et al. Constitutively active calcineurin induces cardiac endoplasmic reticulum stress and protects against apoptosis that is mediated by alpha-crystallin-B. Proc Natl Acad Sci U S A. 2010;107:18481–18486. - PMC - PubMed
-
- Dawson MA, Kouzarides T, Huntly BJ. Targeting epigenetic readers in cancer. N Engl J Med. 2012;367:647–657. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
