

Automated identification of functional dynamic contact networks from X-ray crystallography
- PMID: 23913260
- PMCID: PMC3760795
- DOI: 10.1038/nmeth.2592
Automated identification of functional dynamic contact networks from X-ray crystallography
Abstract
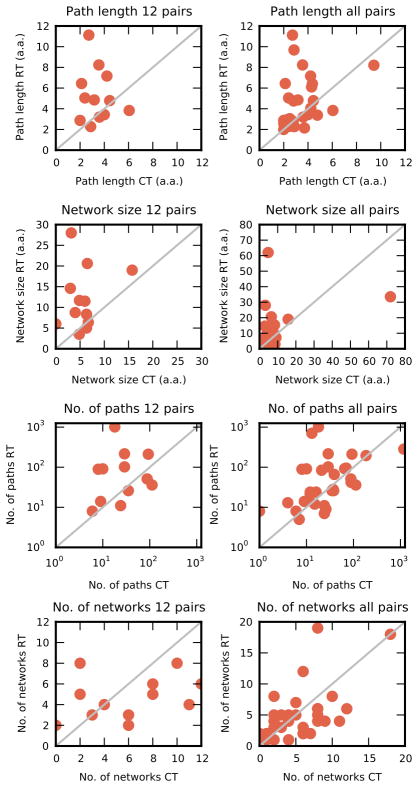
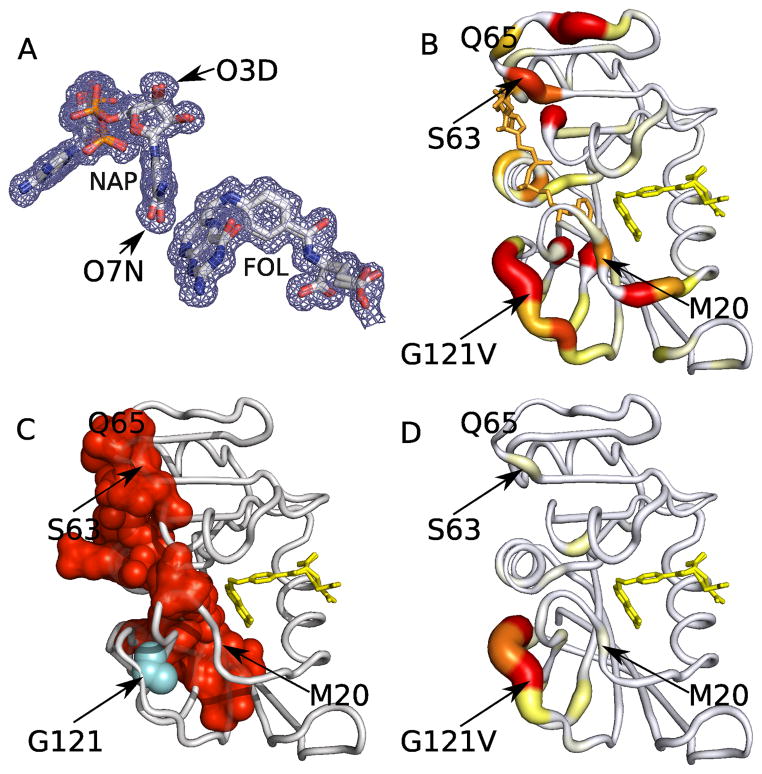
Protein function often depends on the exchange between conformational substates. Allosteric ligand binding or distal mutations can stabilize specific active-site conformations and consequently alter protein function. Observing alternative conformations at low levels of electron density, in addition to comparison of independently determined X-ray crystal structures, can provide mechanistic insights into conformational dynamics. Here we report a new algorithm, CONTACT, that identifies contact networks of conformationally heterogeneous residues directly from high-resolution X-ray crystallography data. Contact networks determined for Escherichia coli dihydrofolate reductase (ecDHFR) predict the observed long-range pattern of NMR chemical shift perturbations of an allosteric mutation. A comparison of contact networks in wild-type and mutant ecDHFR suggests that mutations that alter optimized contact networks of coordinated motions can impair catalytic function. CONTACT-guided mutagenesis can exploit the structure-dynamics-function relationship in protein engineering and design.
Conflict of interest statement
The authors declare no competing financial interests.
Figures



Comment in
-
Visualizing networks of mobility in proteins.Nat Methods. 2013 Sep;10(9):835-7. doi: 10.1038/nmeth.2606. Nat Methods. 2013. PMID: 23985728 No abstract available.
References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
