

Profibrotic TGFβ responses require the cooperative action of PDGF and ErbB receptor tyrosine kinases
- PMID: 23913859
- PMCID: PMC3804746
- DOI: 10.1096/fj.12-224907
Profibrotic TGFβ responses require the cooperative action of PDGF and ErbB receptor tyrosine kinases
Abstract
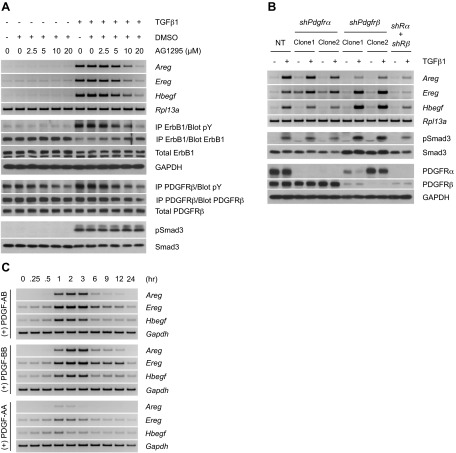
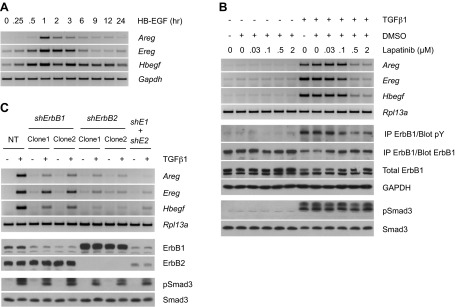
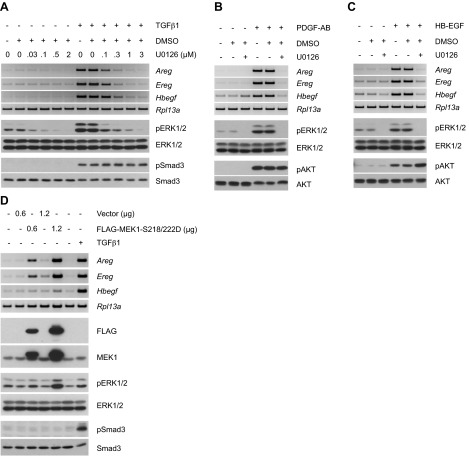
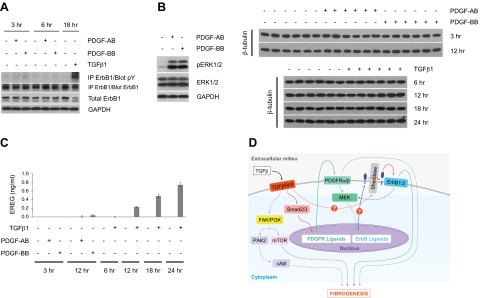
Transforming growth factor β (TGFβ) has significant profibrotic activity both in vitro and in vivo. This reflects its capacity to stimulate fibrogenic mediators and induce the expression of other profibrotic cytokines such as platelet-derived growth factor (PDGF) and epidermal growth factor (EGF/ErbB) ligands. Here we address both the mechanisms by which TGFβ induced ErbB ligands and the physiological significance of inhibiting multiple TGFβ-regulated processes. The data document that ErbB ligand induction requires PDGF receptor (PDGFR) mediation and engages a positive autocrine/paracrine feedback loop via ErbB receptors. Whereas PDGFRs are essential for TGFβ-stimulated ErbB ligand up-regulation, TGFβ-specific signals are also required for ErbB receptor activation. Subsequent profibrotic responses are shown to involve the cooperative action of PDGF and ErbB signaling. Moreover, using a murine treatment model of bleomycin-induced pulmonary fibrosis we found that inhibition of TGFβ/PDGF and ErbB pathways with imatinib plus lapatinib, respectively, not only prevented myofibroblast gene expression to a greater extent than either drug alone, but also essentially stabilized gas exchange (oxygen saturation) as an overall measure of lung function. These observations provide important mechanistic insights into profibrotic TGFβ signaling and indicate that targeting multiple cytokines represents a possible strategy to ameliorate organ fibrosis dependent on TGFβ.
Keywords: EGF; pulmonary fibrosis.
Figures
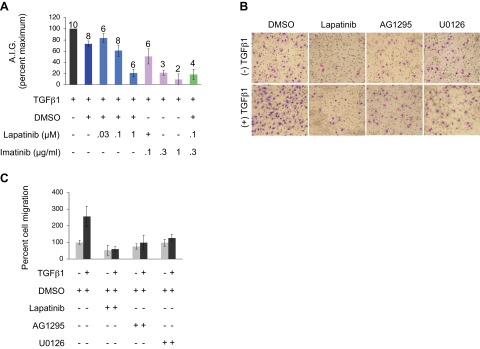
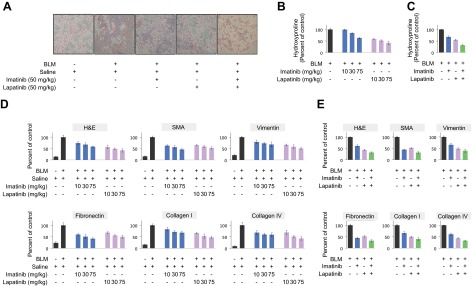
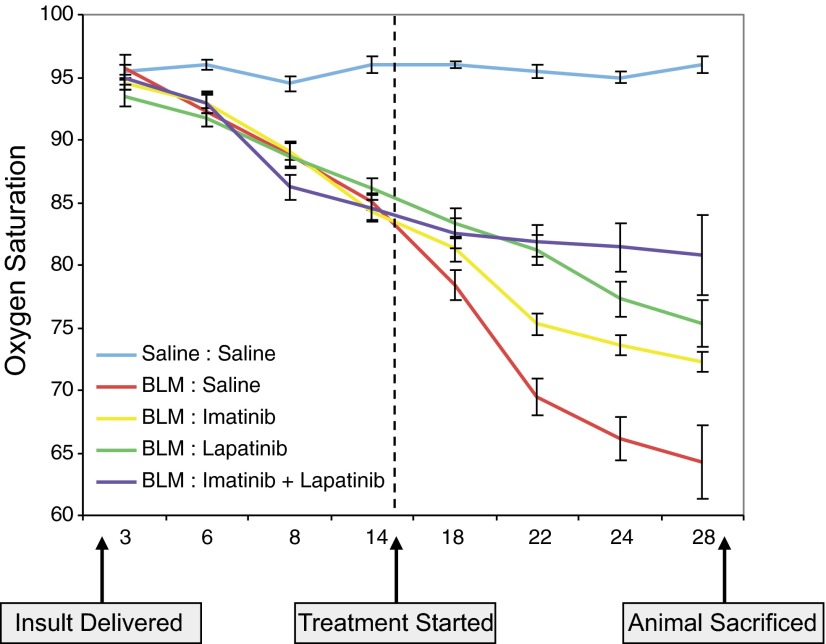
References
-
- Steele M. P., Schwartz D. A. (2012) Molecular mechanisms in progressive idiopathic pulmonary fibrosis. Annu. Rev. Med. 64, 265–276 - PubMed
-
- Sivakumar P., Ntolios P., Jenkins G., Laurent G. (2012) Into the matrix: targeting fibroblasts in pulmonary fibrosis. Curr. Opin. Pulm. Med. 18, 462–469 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
