

Efficient and Unbiased Sampling of Biomolecular Systems in the Canonical Ensemble: A Review of Self-Guided Langevin Dynamics
- PMID: 23913991
- PMCID: PMC3731171
- DOI: 10.1002/9781118197714.ch6
Efficient and Unbiased Sampling of Biomolecular Systems in the Canonical Ensemble: A Review of Self-Guided Langevin Dynamics
Abstract
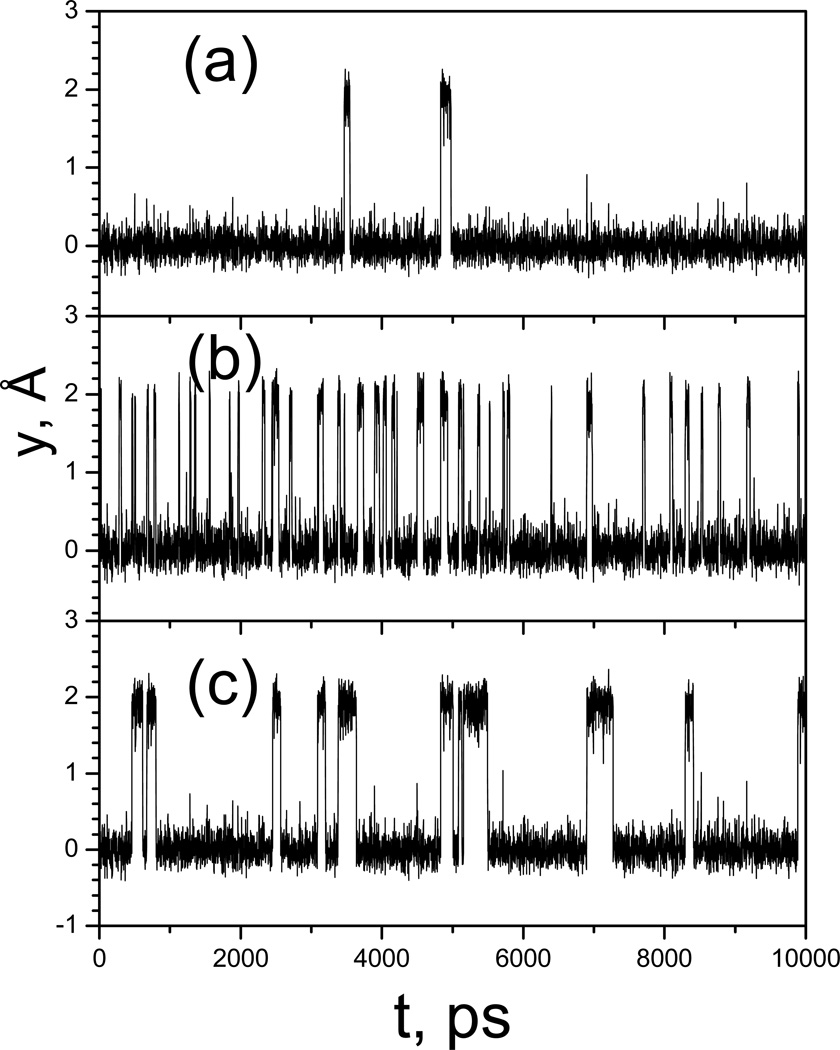
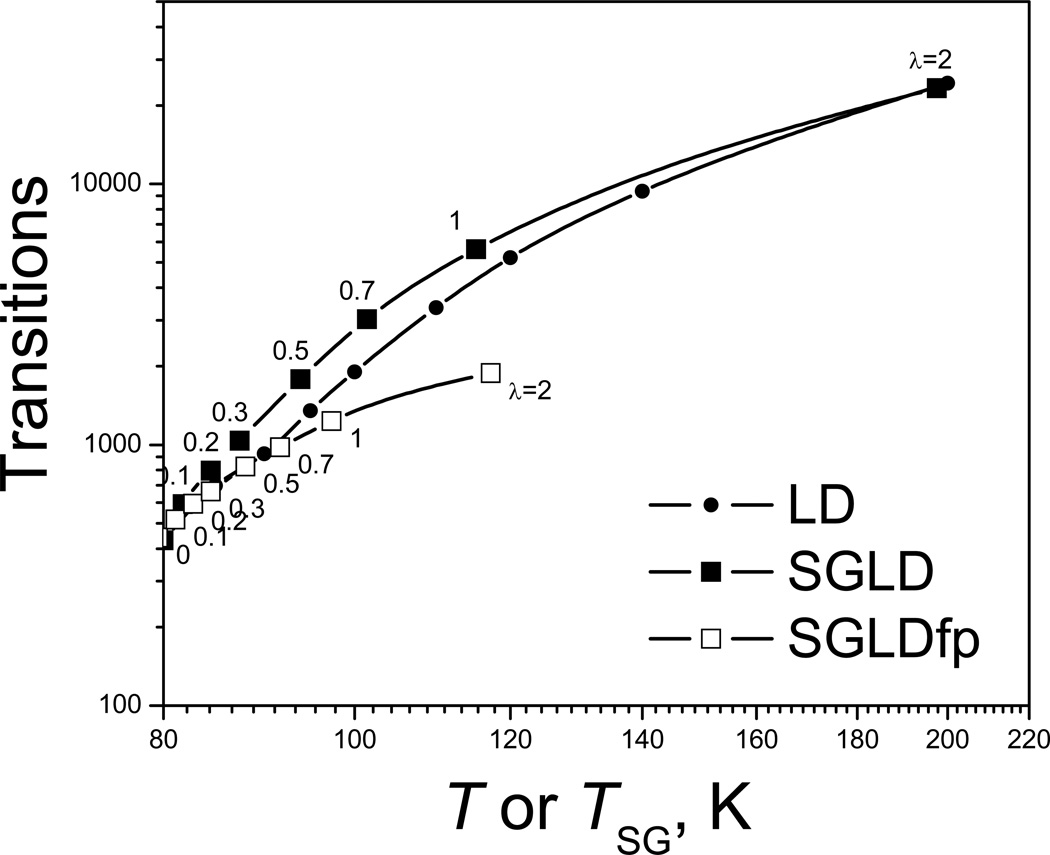
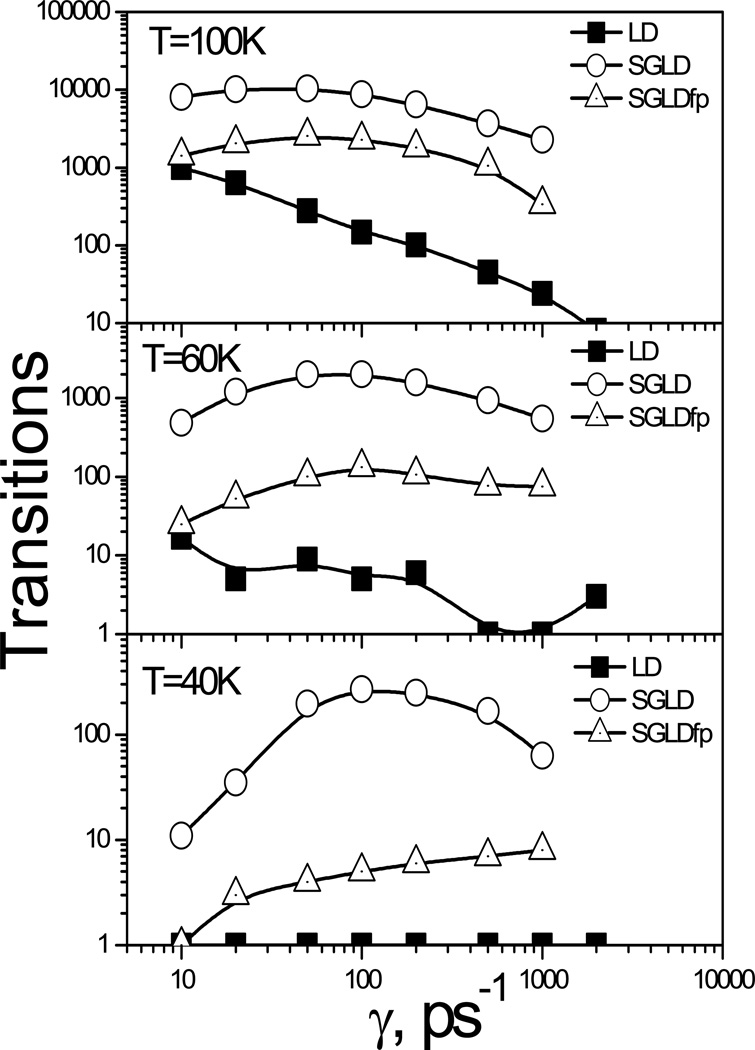
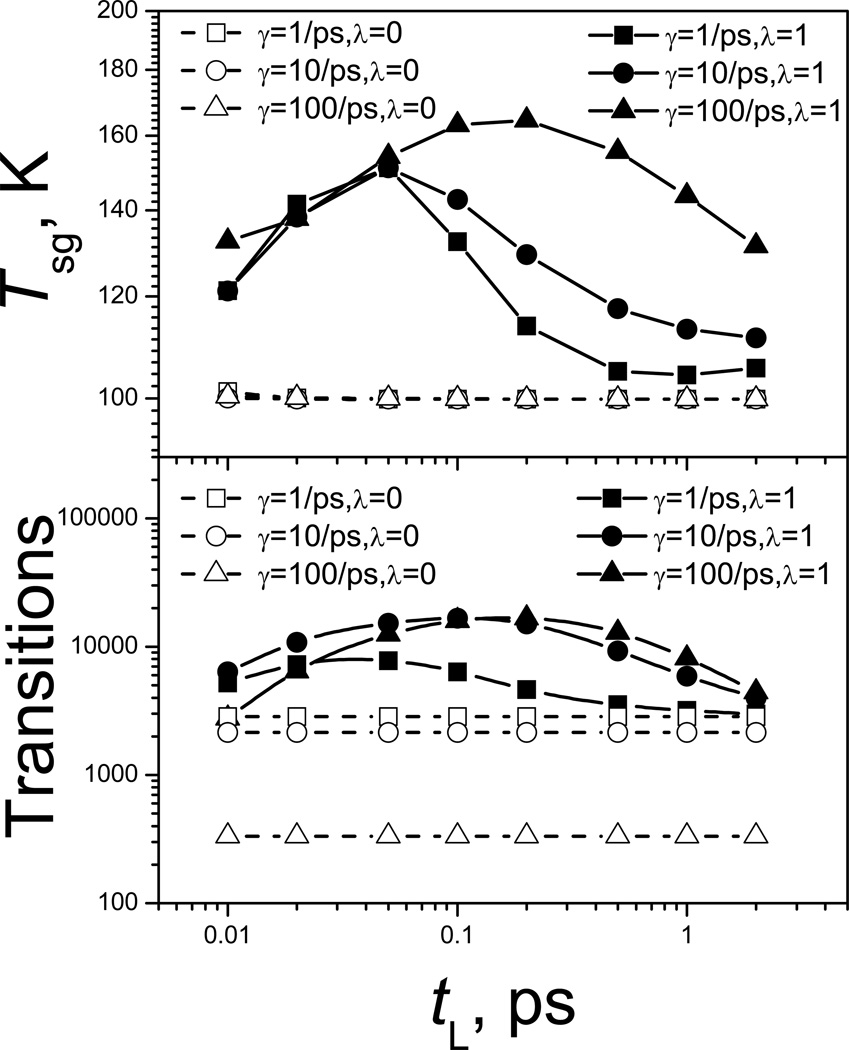
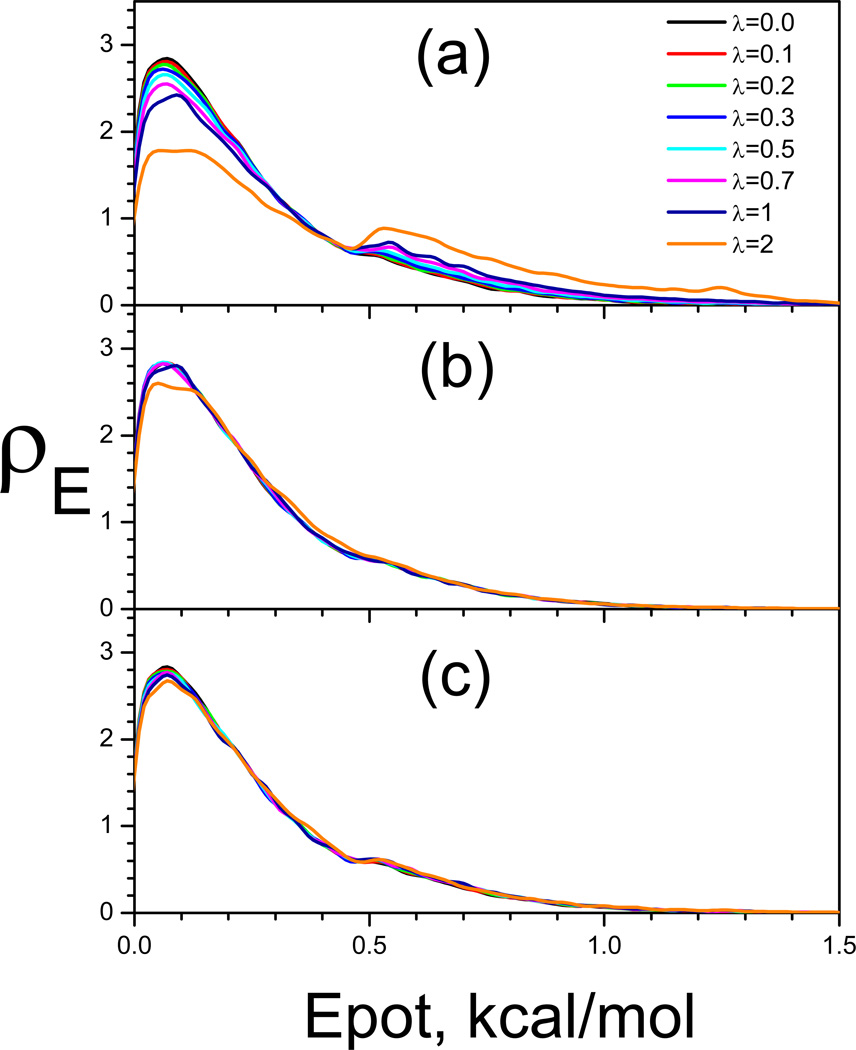
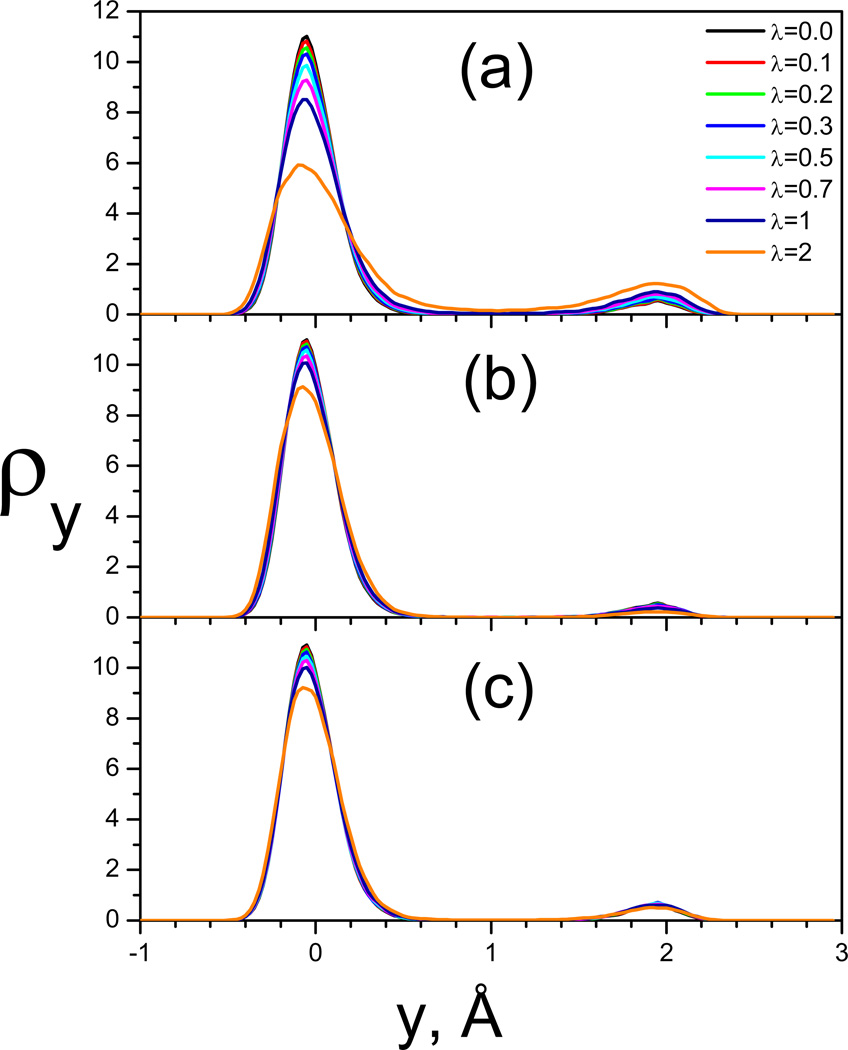
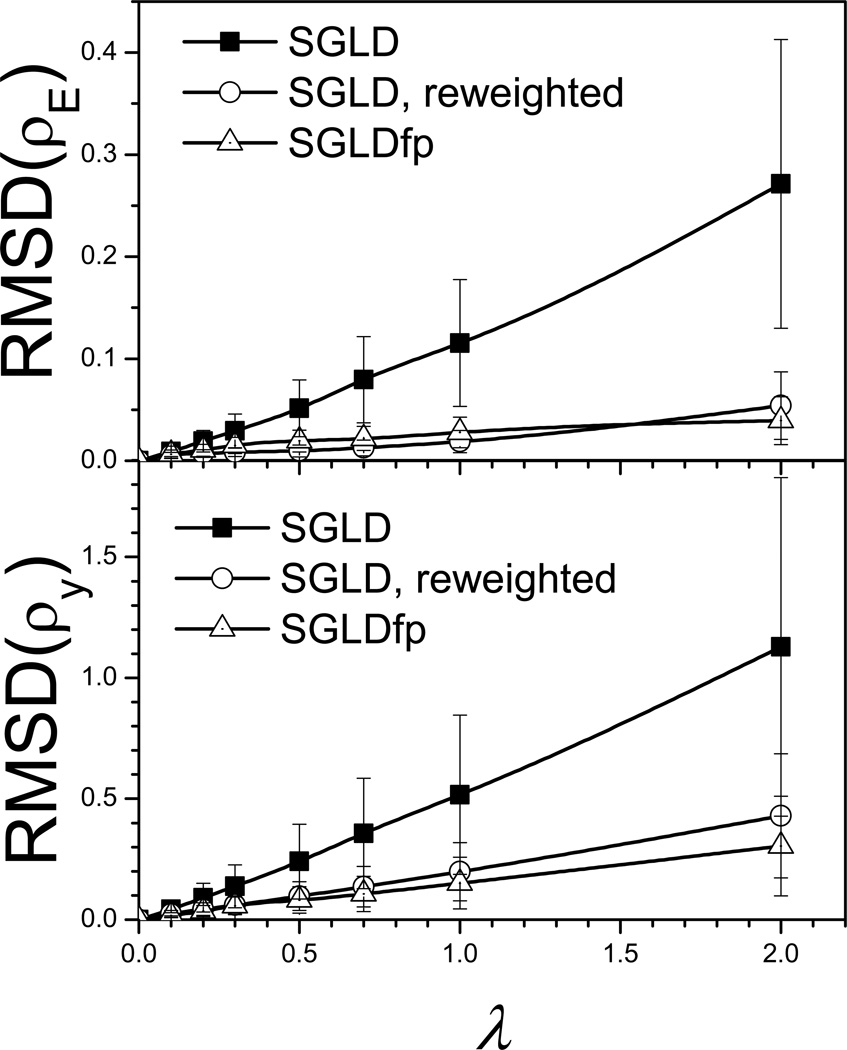
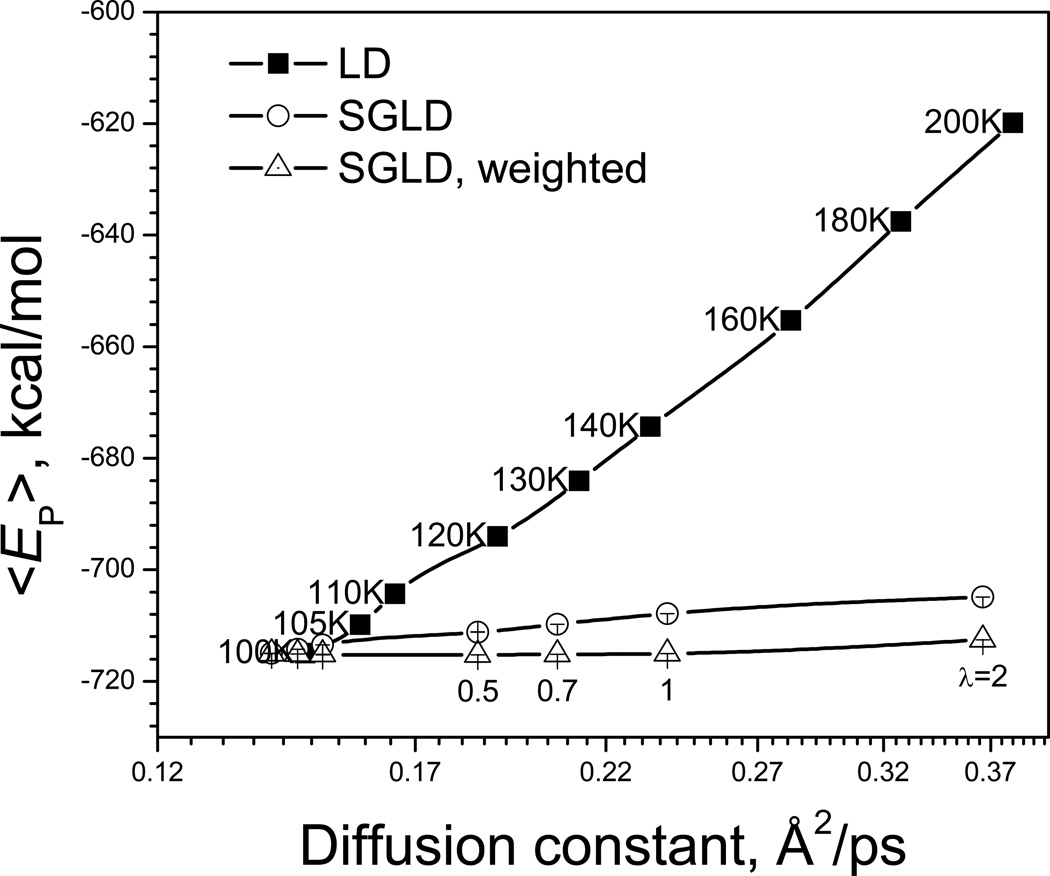
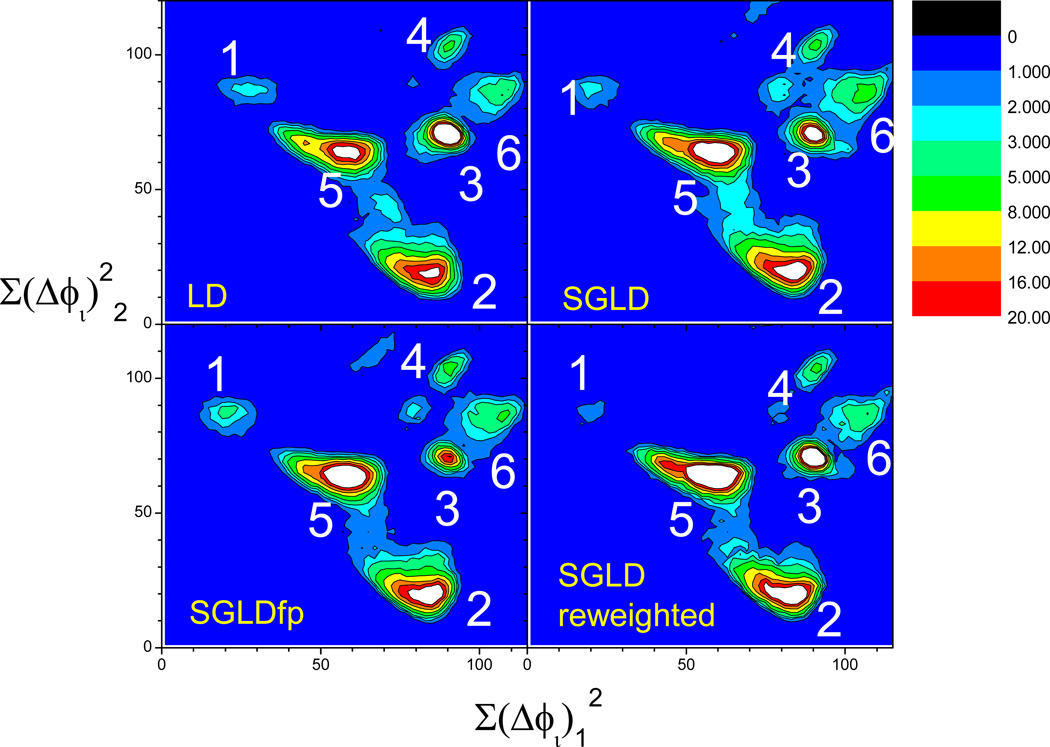
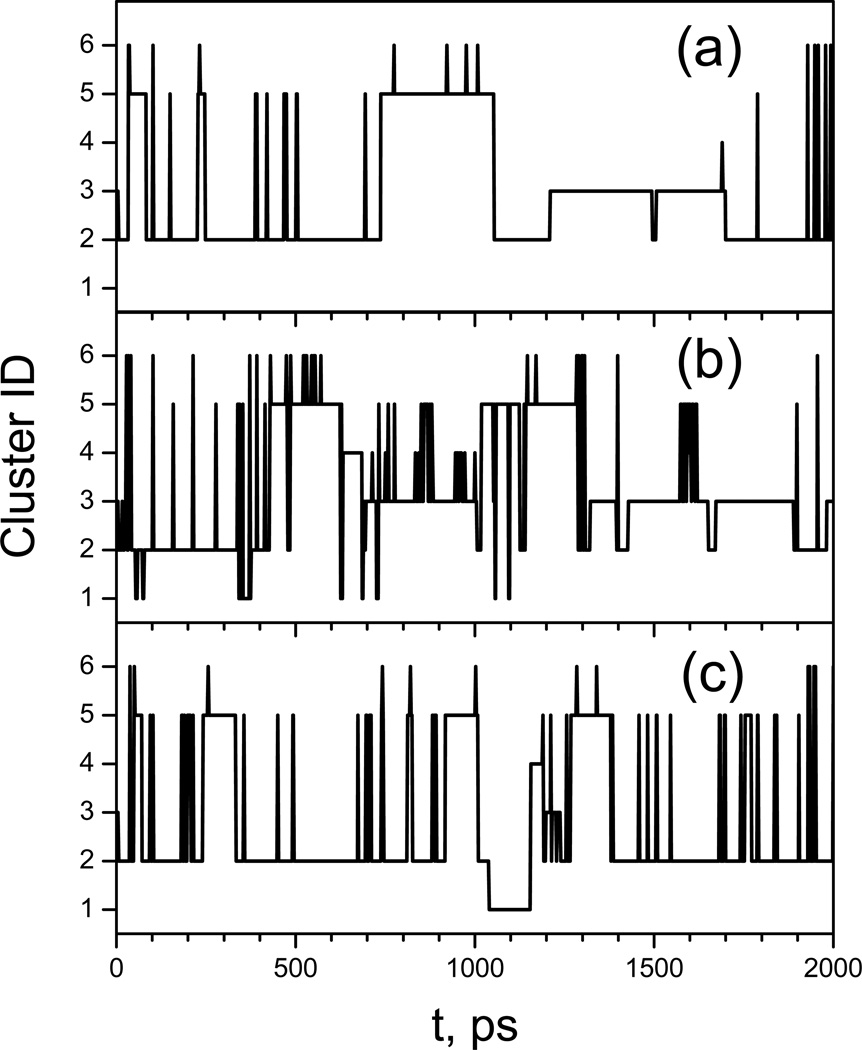
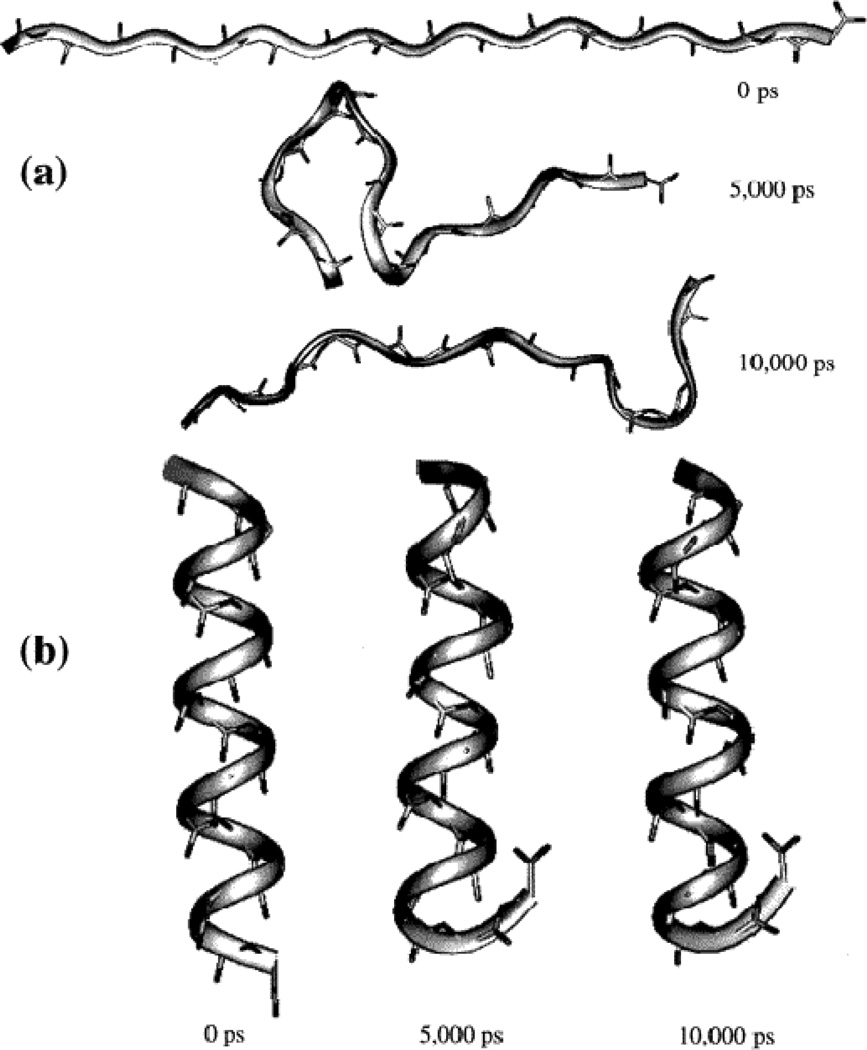
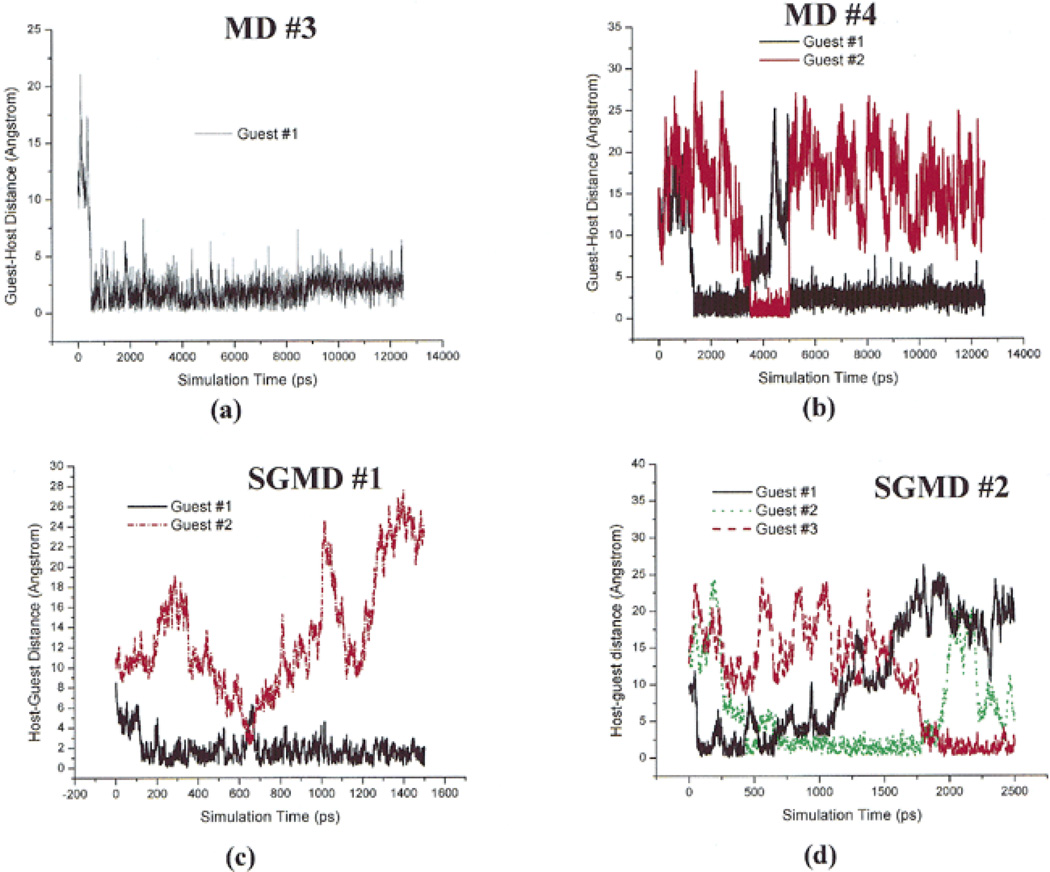
This review provides a comprehensive description of the self-guided Langevin dynamics (SGLD) and the self-guided molecular dynamics (SGMD) methods and their applications. Example systems are included to provide guidance on optimal application of these methods in simulation studies. SGMD/SGLD has enhanced ability to overcome energy barriers and accelerate rare events to affordable time scales. It has been demonstrated that with moderate parameters, SGLD can routinely cross energy barriers of 20 kT at a rate that molecular dynamics (MD) or Langevin dynamics (LD) crosses 10 kT barriers. The core of these methods is the use of local averages of forces and momenta in a direct manner that can preserve the canonical ensemble. The use of such local averages results in methods where low frequency motion "borrows" energy from high frequency degrees of freedom when a barrier is approached and then returns that excess energy after a barrier is crossed. This self-guiding effect also results in an accelerated diffusion to enhance conformational sampling efficiency. The resulting ensemble with SGLD deviates in a small way from the canonical ensemble, and that deviation can be corrected with either an on-the-fly or a post processing reweighting procedure that provides an excellent canonical ensemble for systems with a limited number of accelerated degrees of freedom. Since reweighting procedures are generally not size extensive, a newer method, SGLDfp, uses local averages of both momenta and forces to preserve the ensemble without reweighting. The SGLDfp approach is size extensive and can be used to accelerate low frequency motion in large systems, or in systems with explicit solvent where solvent diffusion is also to be enhanced. Since these methods are direct and straightforward, they can be used in conjunction with many other sampling methods or free energy methods by simply replacing the integration of degrees of freedom that are normally sampled by MD or LD.
Figures
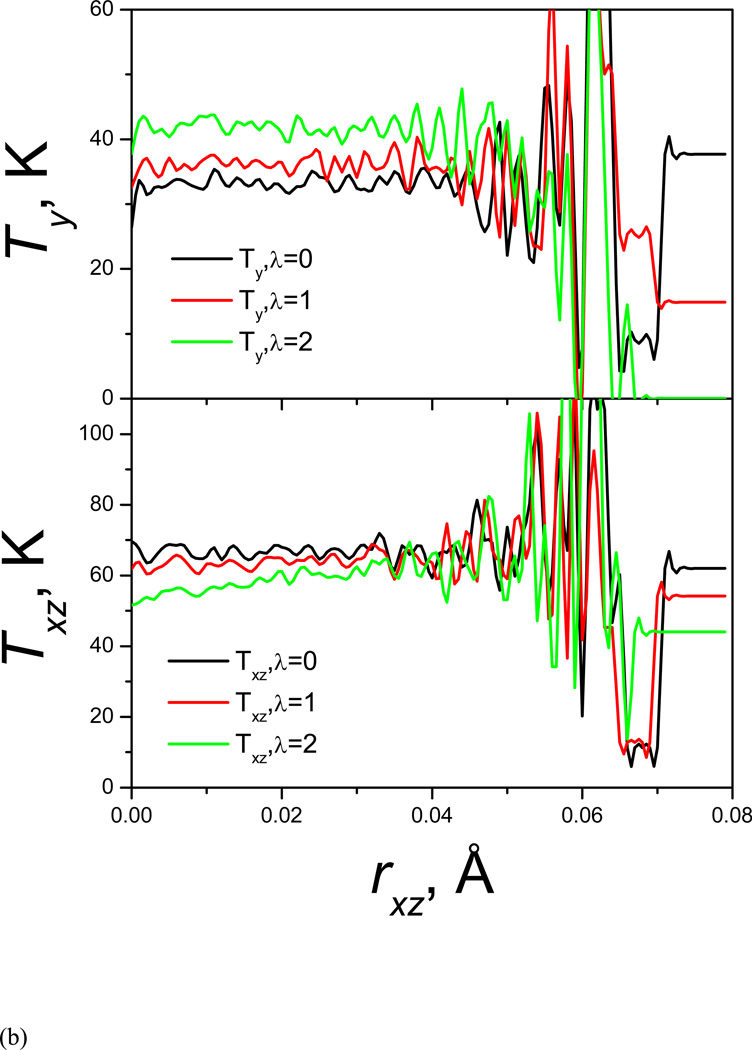
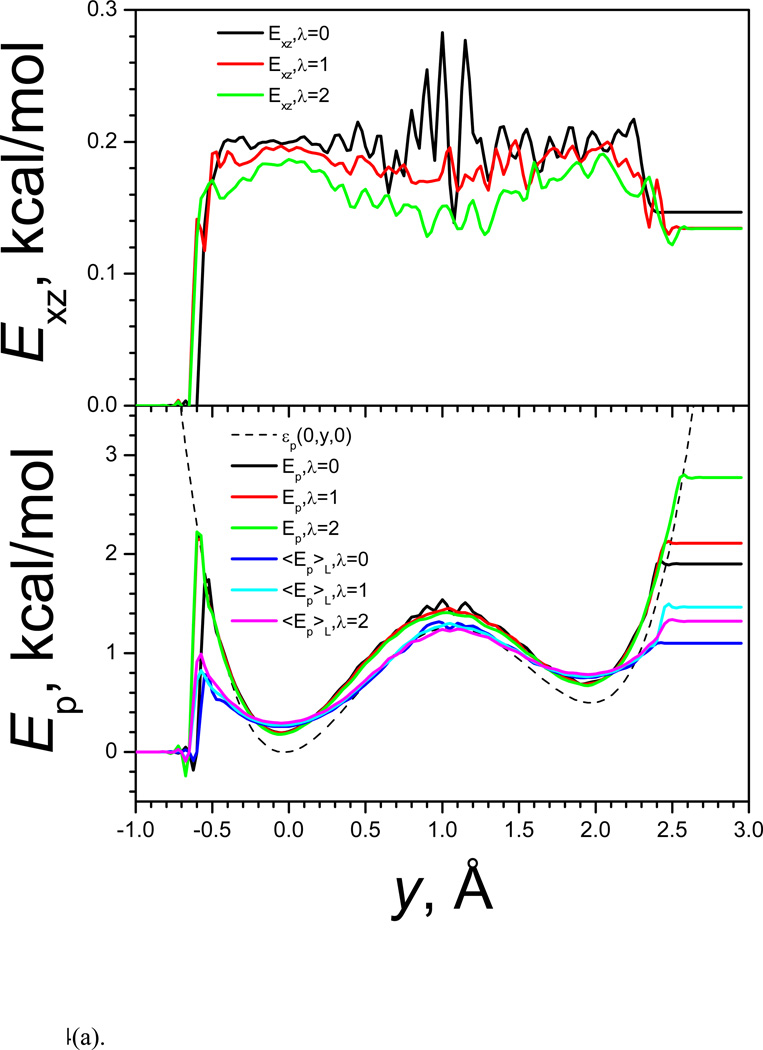
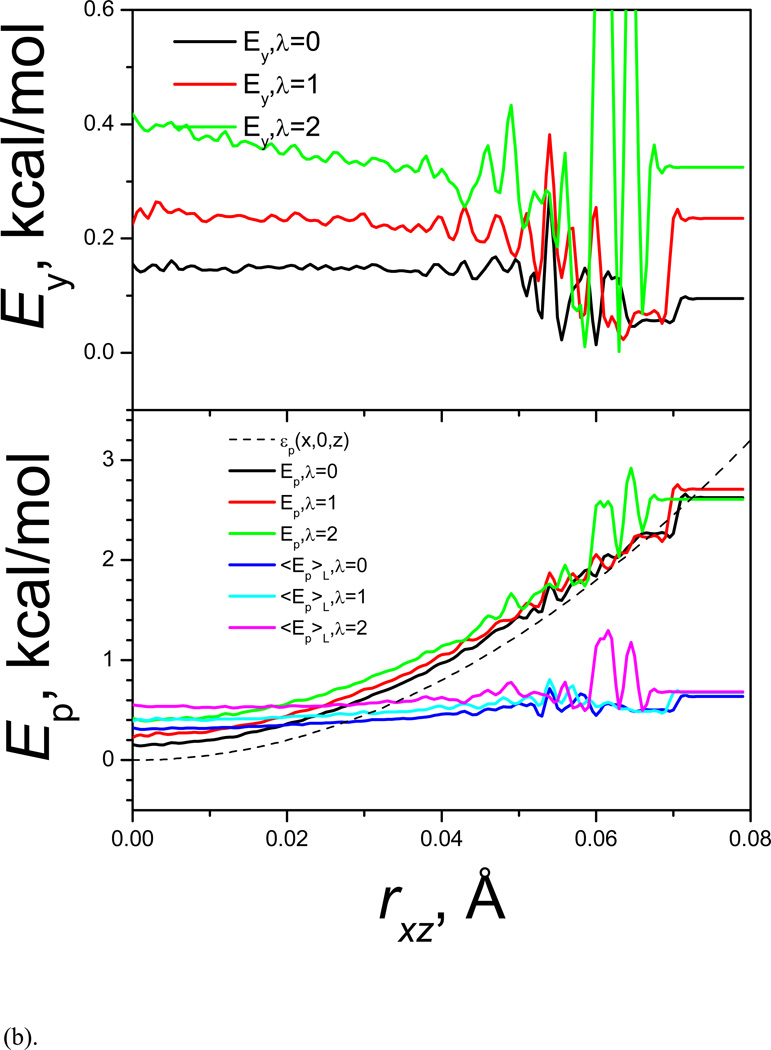
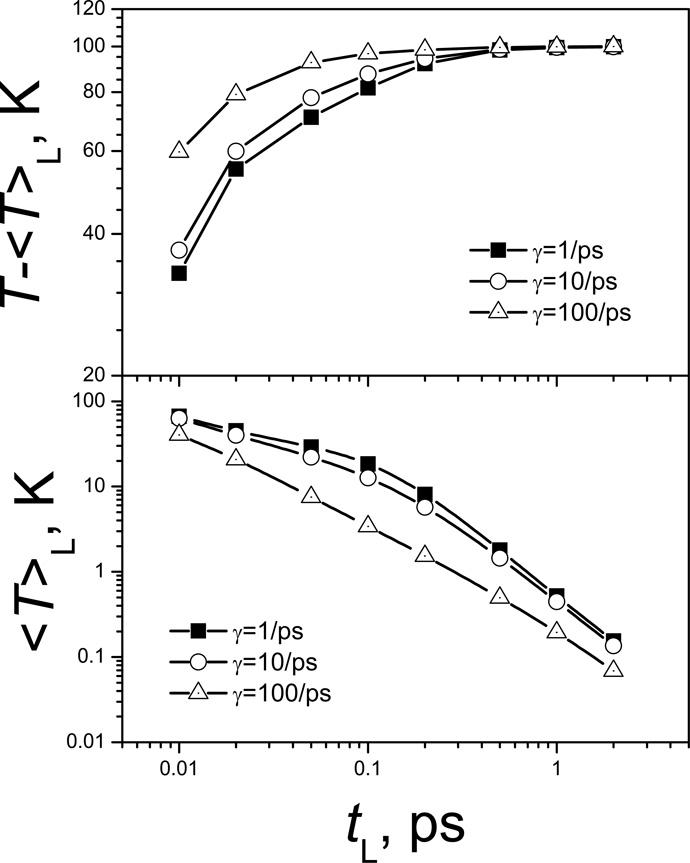


References
-
- Abe Y, Jitsukawa S. Phase transformation of Cu precipitate in Fe-Cu alloy studied using self-guided molecular dynamics. Philosophical Magazine Letters. 2009;89:535–543.
-
- Allen MP, Tildesley DJ. Computer Simulations of Liquids. Oxford: Clarendon Press; 1987.
-
- Andricioaei I, Dinner AR, Karplus M. Self-guided enhanced sampling methods for thermodynamic averages. J. Chem. Phys. 2003;118:1074–1084.
-
- Beckers ML, Buydens LM, Pikkemaat JA, Altona C. Application of a genetic algorithm in the conformational analysis of methylene-acetal-linked thymine dimers in DNA: comparison with distance geometry calculations. J Biomol NMR. 1997;9:25–34. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials