

A comprehensive metatranscriptome analysis pipeline and its validation using human small intestine microbiota datasets
- PMID: 23915218
- PMCID: PMC3750648
- DOI: 10.1186/1471-2164-14-530
A comprehensive metatranscriptome analysis pipeline and its validation using human small intestine microbiota datasets
Abstract
Background: Next generation sequencing (NGS) technologies can be applied in complex microbial ecosystems for metatranscriptome analysis by employing direct cDNA sequencing, which is known as RNA sequencing (RNA-seq). RNA-seq generates large datasets of great complexity, the comprehensive interpretation of which requires a reliable bioinformatic pipeline. In this study, we focus on the development of such a metatranscriptome pipeline, which we validate using Illumina RNA-seq datasets derived from the small intestine microbiota of two individuals with an ileostomy.
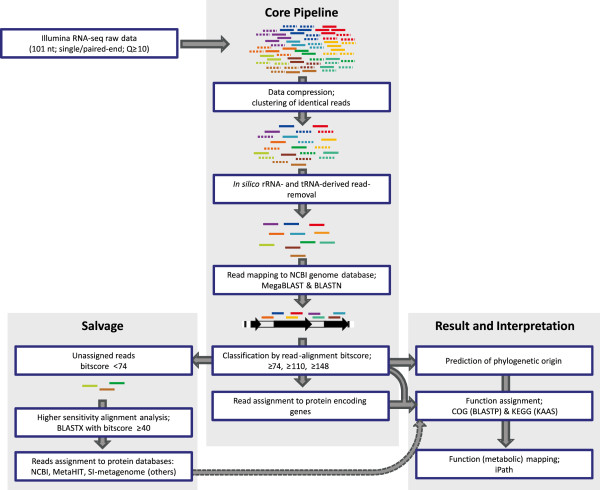
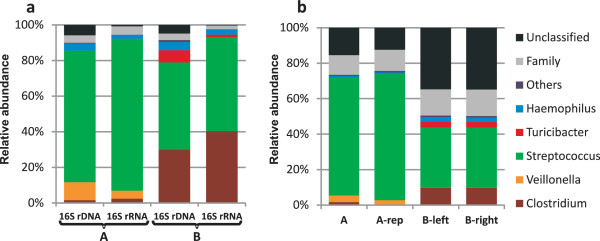
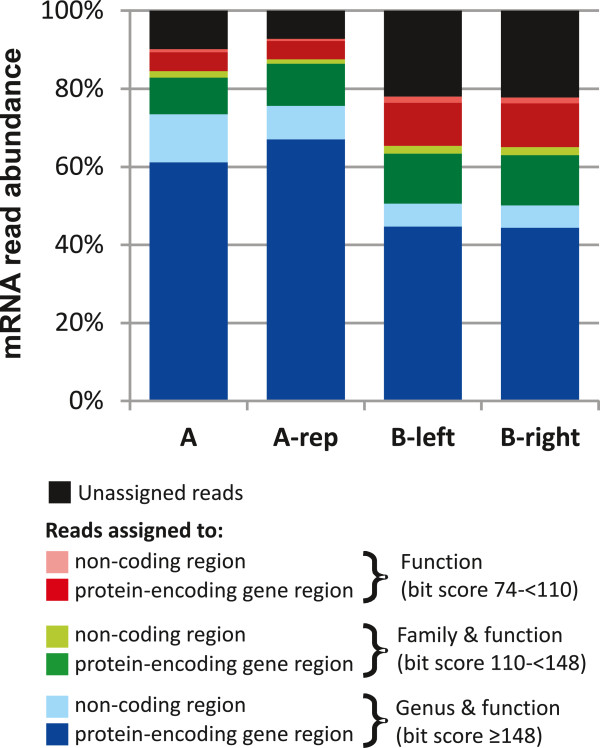
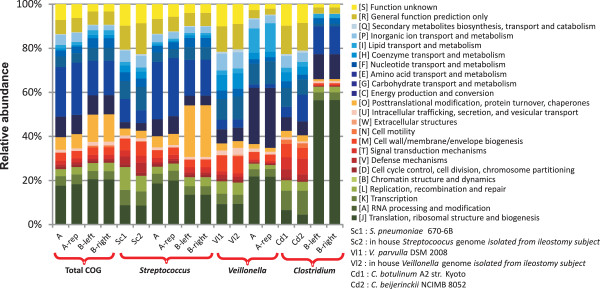
Results: The metatranscriptome pipeline developed here enabled effective removal of rRNA derived sequences, followed by confident assignment of the predicted function and taxonomic origin of the mRNA reads. Phylogenetic analysis of the small intestine metatranscriptome datasets revealed a strong similarity with the community composition profiles obtained from 16S rDNA and rRNA pyrosequencing, indicating considerable congruency between community composition (rDNA), and the taxonomic distribution of overall (rRNA) and specific (mRNA) activity among its microbial members. Reproducibility of the metatranscriptome sequencing approach was established by independent duplicate experiments. In addition, comparison of metatranscriptome analysis employing single- or paired-end sequencing methods indicated that the latter approach does not provide improved functional or phylogenetic insights. Metatranscriptome functional-mapping allowed the analysis of global, and genus specific activity of the microbiota, and illustrated the potential of these approaches to unravel syntrophic interactions in microbial ecosystems.
Conclusions: A reliable pipeline for metatransciptome data analysis was developed and evaluated using RNA-seq datasets obtained for the human small intestine microbiota. The set-up of the pipeline is very generic and can be applied for (bacterial) metatranscriptome analysis in any chosen niche.
Figures
References
-
- van den Bogert B, Leimena MM, de Vos WM, Zoetendal EG, Kleerebezem M. In: Handbook of Molecular Microbial Ecology. Volume 2. de Bruin FJ, editor. Hoboken, New Jersey: Wiley-Blackwell; 2011. Functional Intestinal Metagenomics; pp. 170–190.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
