

How old is my gene?
- PMID: 23915718
- PMCID: PMC3812327
- DOI: 10.1016/j.tig.2013.07.001
How old is my gene?
Abstract
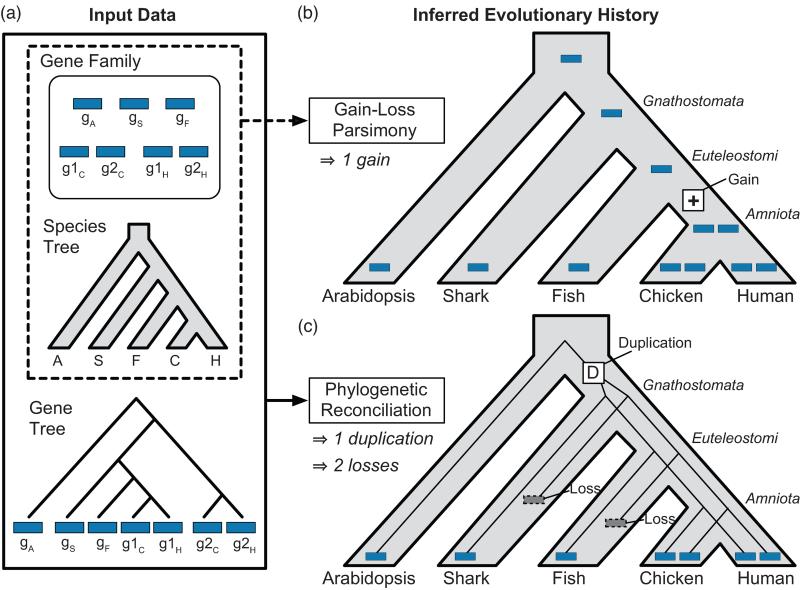
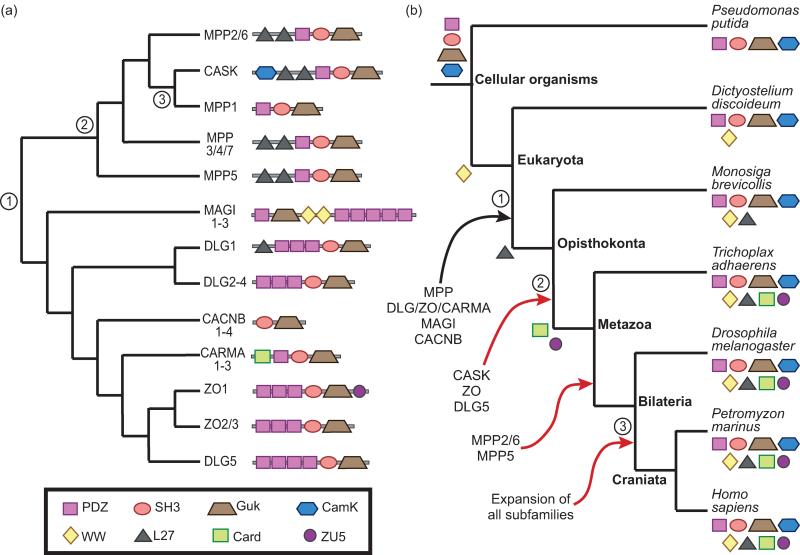
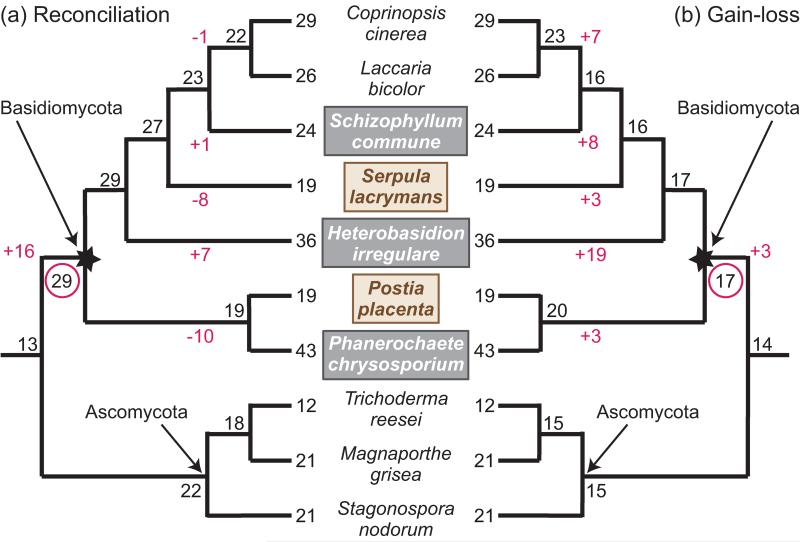
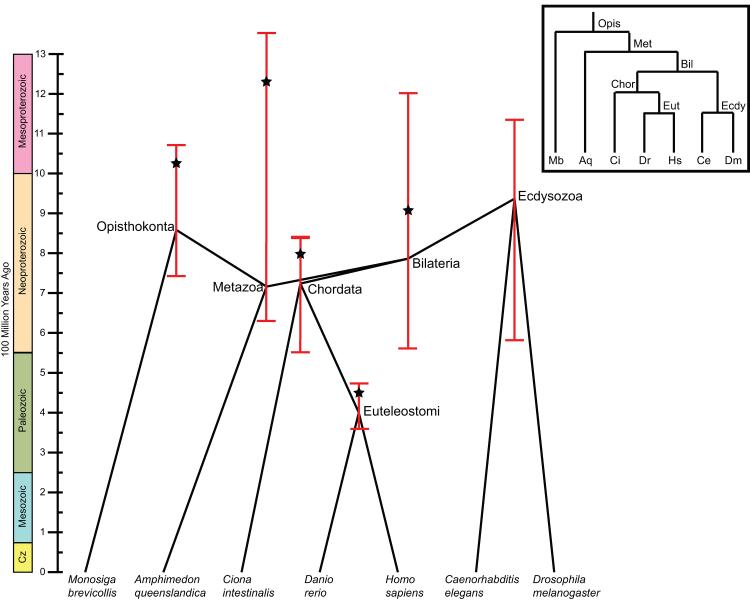
Gene functions, interactions, disease associations, and ecological distributions are all correlated with gene age. However, it is challenging to estimate the intricate series of evolutionary events leading to a modern-day gene and then to reduce this history to a single age estimate. Focusing on eukaryotic gene families, we introduce a framework that can be used to compare current strategies for quantifying gene age, discuss key differences between these methods, and highlight several common problems. We argue that genes with complex evolutionary histories do not have a single well-defined age. As a result, care must be taken to articulate the goals and assumptions of any analysis that uses gene age estimates. Recent algorithmic advances offer the promise of gene age estimates that are fast, accurate, and consistent across gene families. This will enable a shift to integrated genome-wide analyses of all events in gene evolutionary histories in the near future.
Keywords: eukaryotes; gene age; molecular clock; phylogenetics.
Copyright © 2013 Elsevier Ltd. All rights reserved.
Figures

References
-
- Domazet-Loso T, Tautz D. A Phylogenetically Based Transcriptome Age Index Mirrors Ontogenetic Divergence Patterns. Nature. 2010;468:815–8. - PubMed
-
- Alba MM, Castresana J. Inverse Relationship between Evolutionary Rate and Age of Mammalian Genes. Mol. Biol. Evol. 2005;22:598–606. - PubMed
-
- Cai JJ, et al. Accelerated Evolutionary Rate May Be Responsible for the Emergence of Lineage-Specific Genes in Ascomycota. J. Mol. Evol. 2006;63:1–11. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
