

PARP and CHK inhibitors interact to cause DNA damage and cell death in mammary carcinoma cells
- PMID: 23917378
- PMCID: PMC3672190
- DOI: 10.4161/cbt.24424
PARP and CHK inhibitors interact to cause DNA damage and cell death in mammary carcinoma cells
Abstract
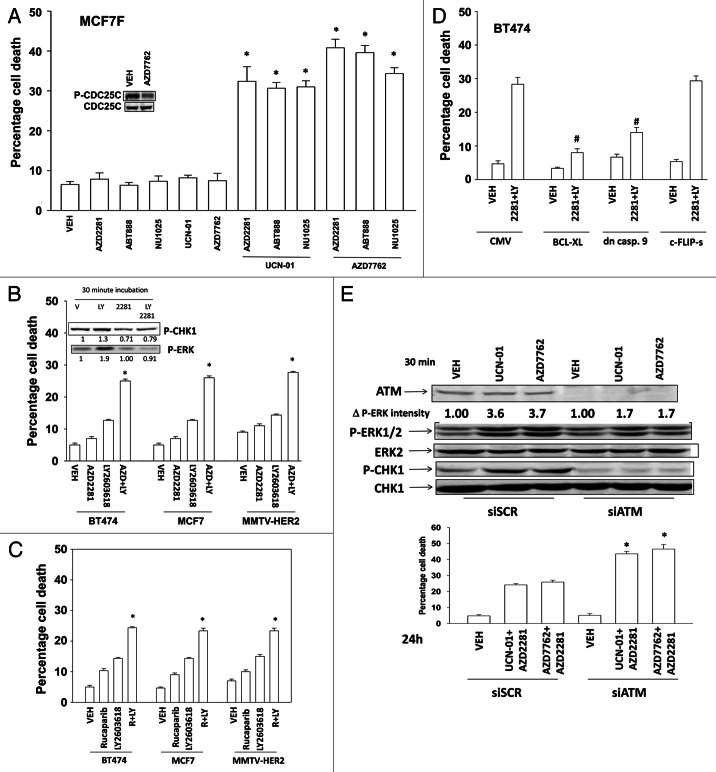
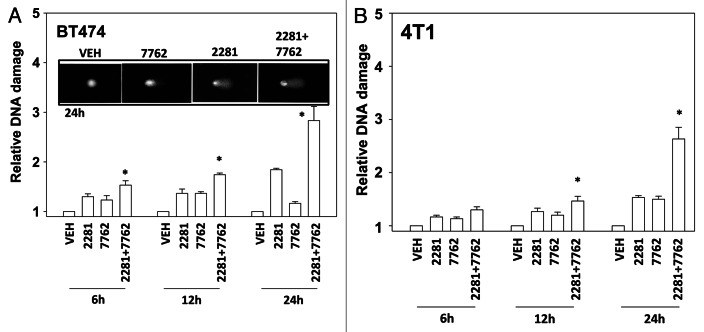
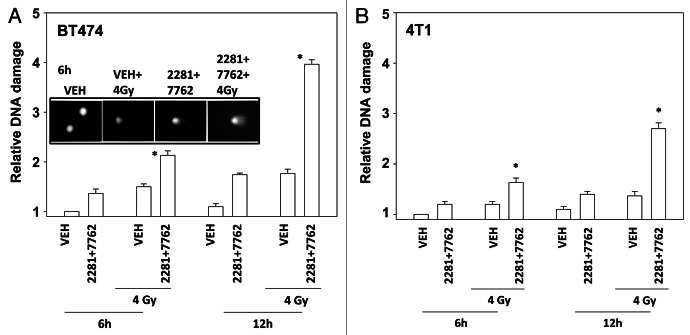
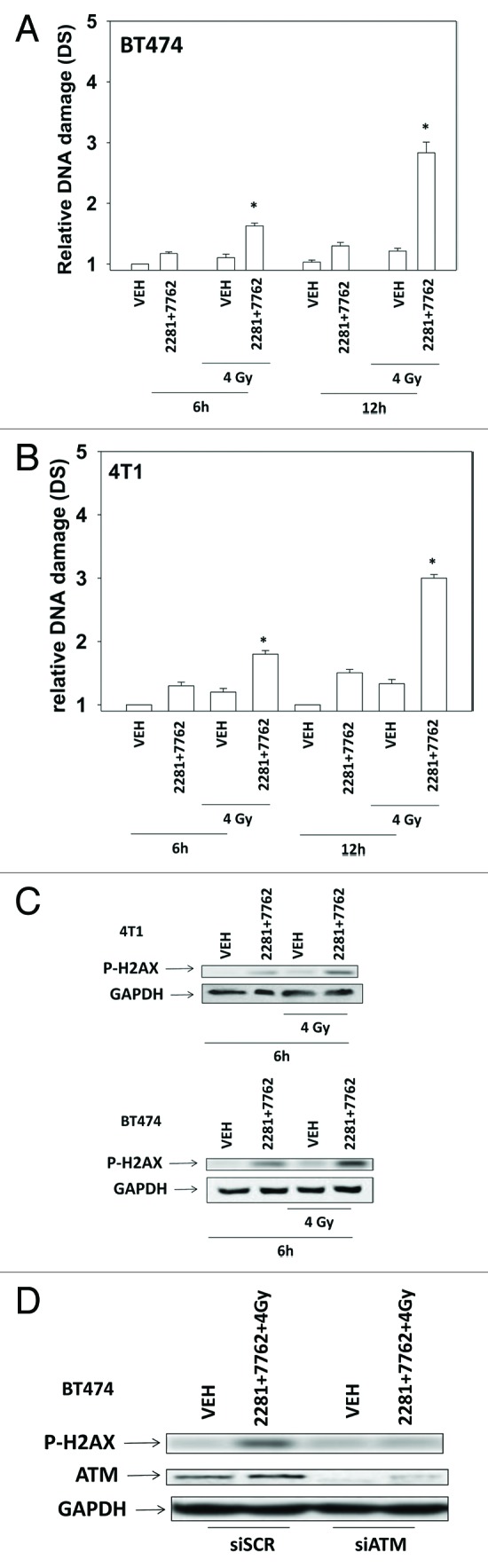
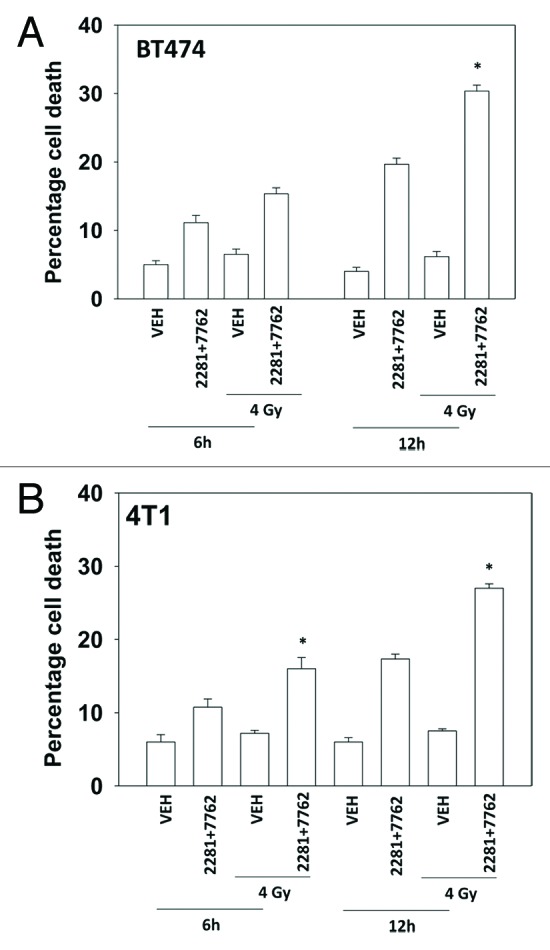
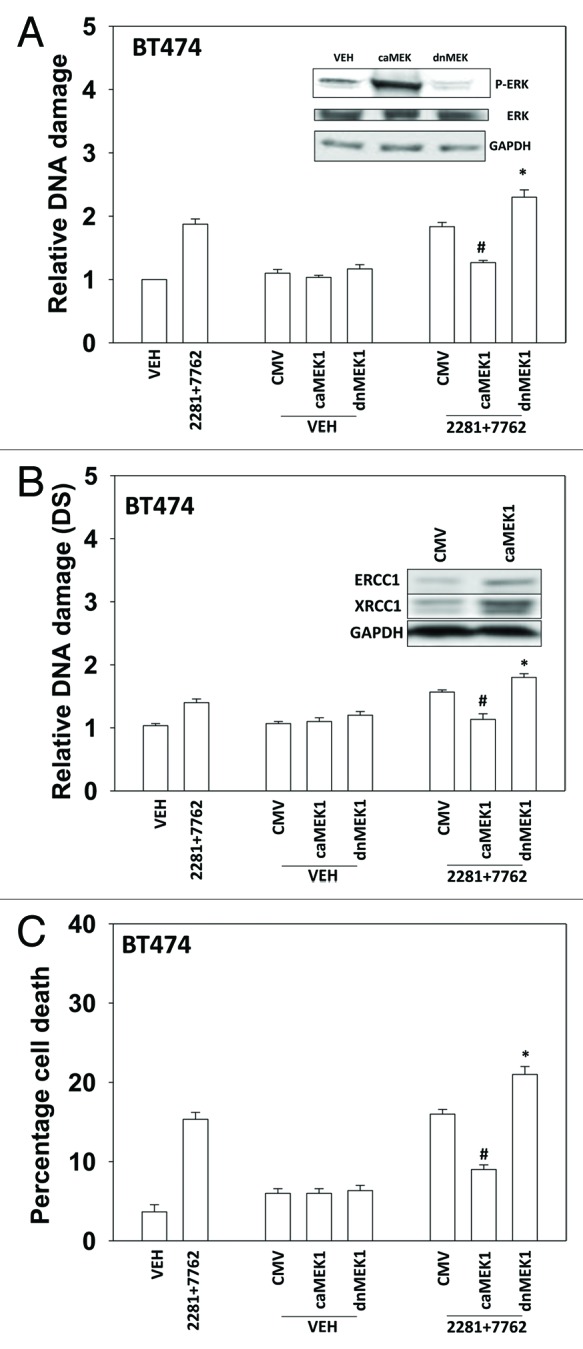
The present studies examined viability and DNA damage levels in mammary carcinoma cells following PARP1 and CHK1 inhibitor drug combination exposure. PARP1 inhibitors [AZD2281 ; ABT888 ; NU1025 ; AG014699] interacted with CHK1 inhibitors [UCN-01 ; AZD7762 ; LY2603618] to kill mammary carcinoma cells. PARP1 and CHK1 inhibitors interacted to increase both single strand and double strand DNA breaks that correlated with increased γH2AX phosphorylation. Treatment of cells with CHK1 inhibitors increased the phosphorylation of CHK1 and ERK1/2. Knock down of ATM suppressed the drug-induced increases in CHK1 and ERK1/2 phosphorylation and enhanced tumor cell killing by PARP1 and CHK1 inhibitors. Expression of dominant negative MEK1 enhanced drug-induced DNA damage whereas expression of activated MEK1 suppressed both the DNA damage response and tumor cell killing. Collectively our data demonstrate that PARP1 and CHK1 inhibitors interact to kill mammary carcinoma cells and that increased DNA damage is a surrogate marker for the response of cells to this drug combination.
Keywords: ATM; CHK1; DNA damage; PARP1; apoptosis; comet; kinase.
Figures
References
-
- Morgan MA, Parsels LA, Zhao L, Parsels JD, Davis MA, Hassan MC, et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 2010;70:4972–81. doi: 10.1158/0008-5472.CAN-09-3573. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous