

The integrated landscape of driver genomic alterations in glioblastoma
- PMID: 23917401
- PMCID: PMC3799953
- DOI: 10.1038/ng.2734
The integrated landscape of driver genomic alterations in glioblastoma
Abstract
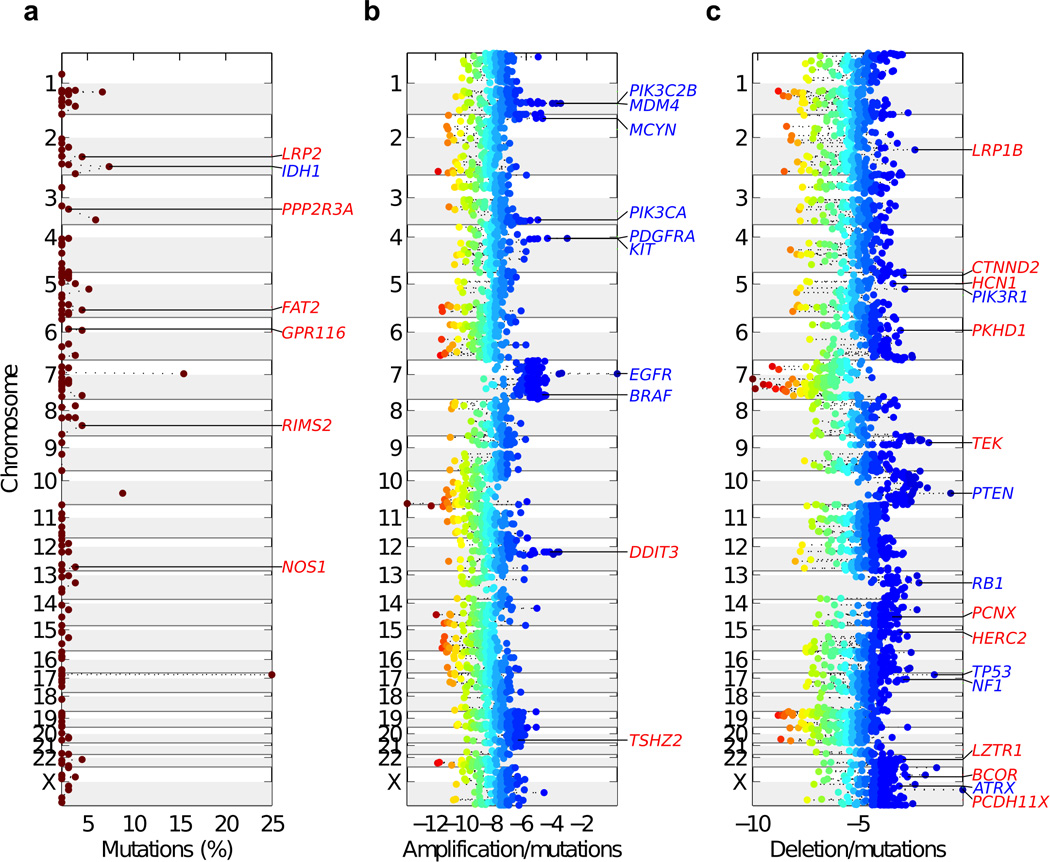
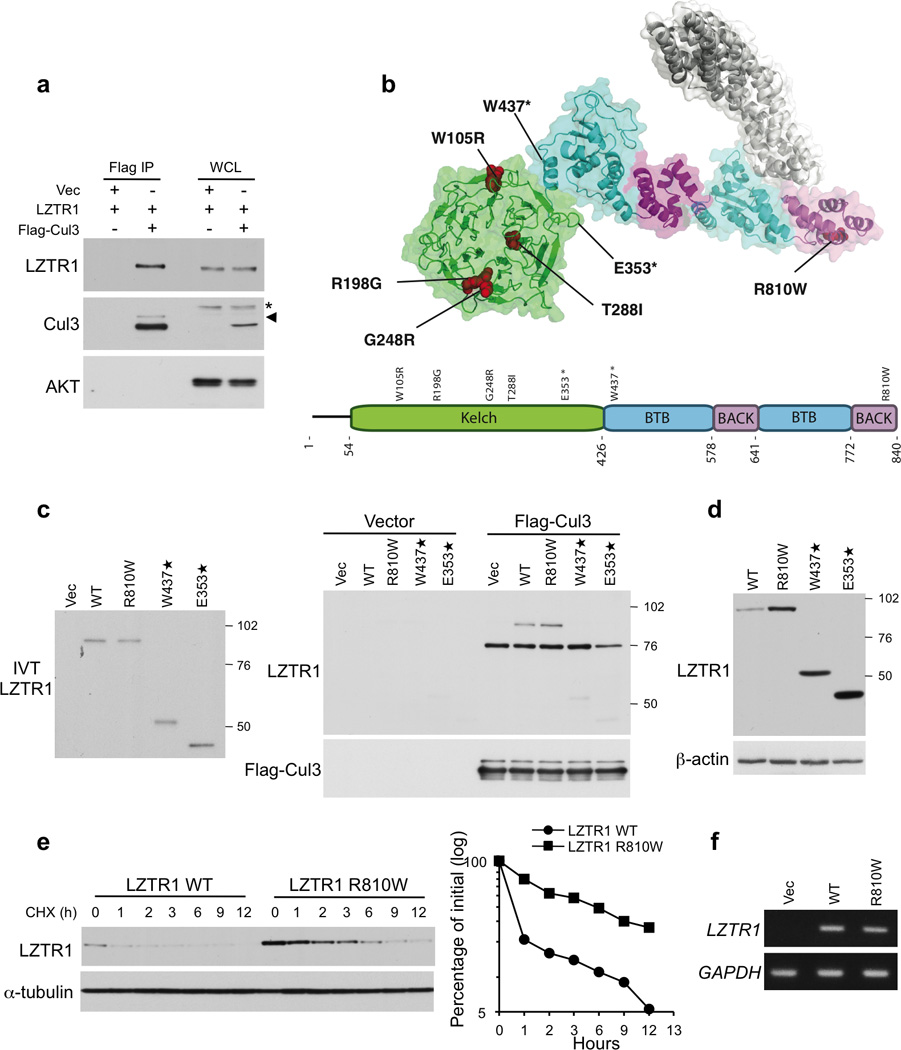
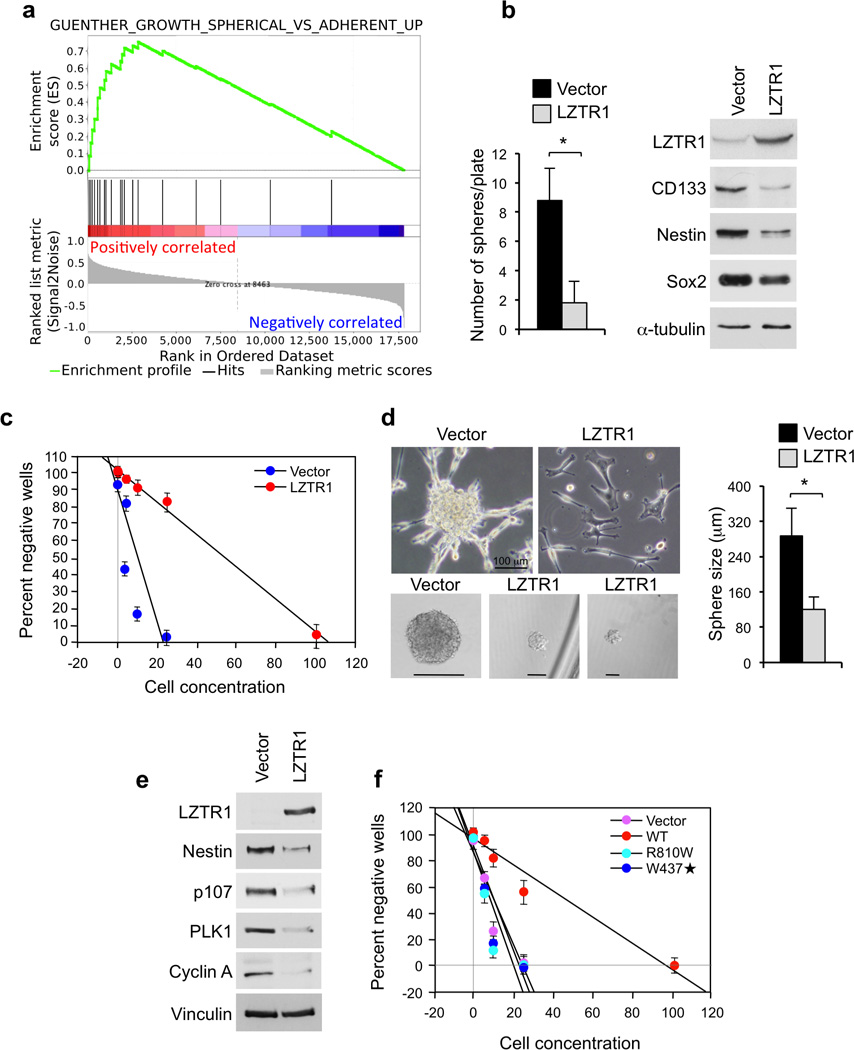
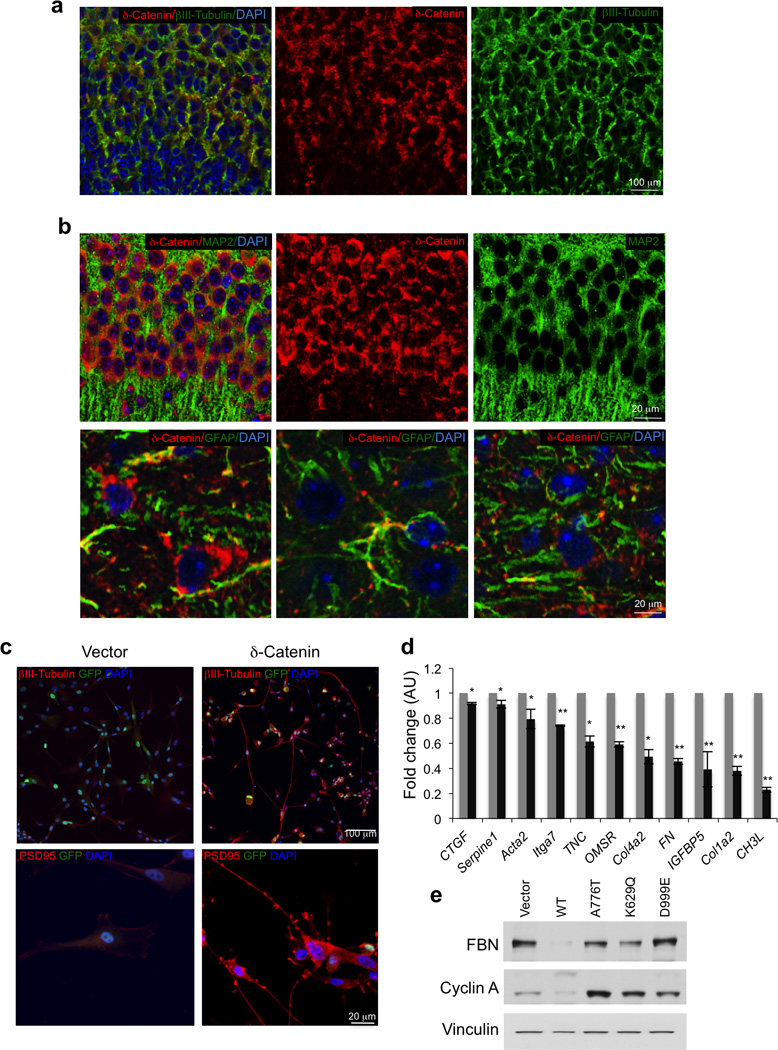
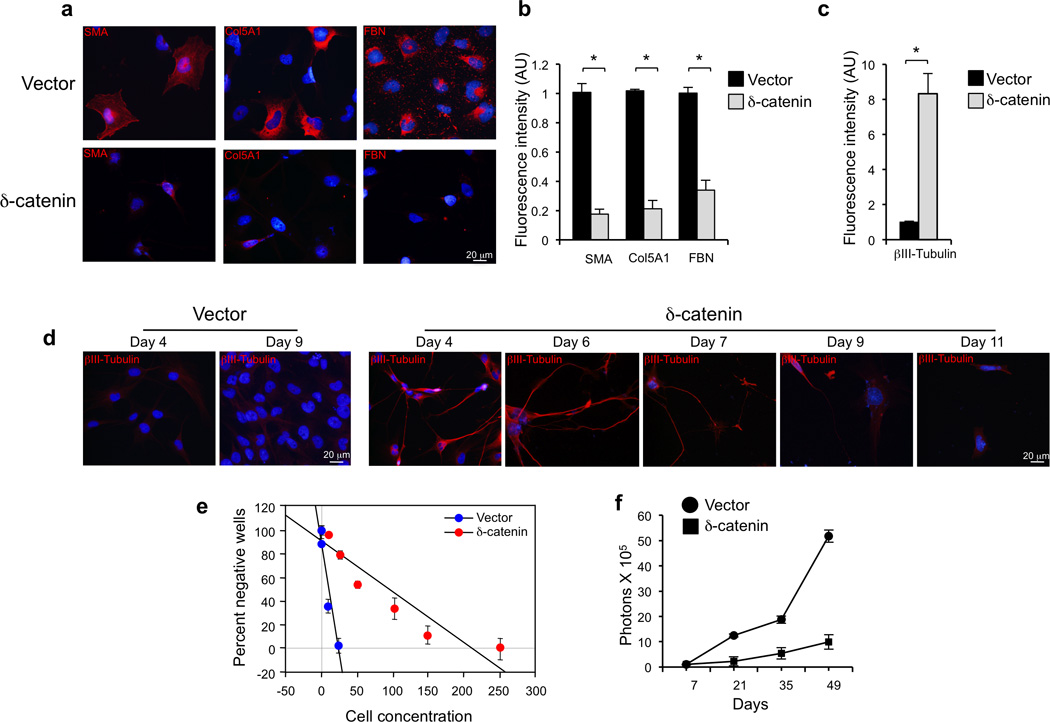
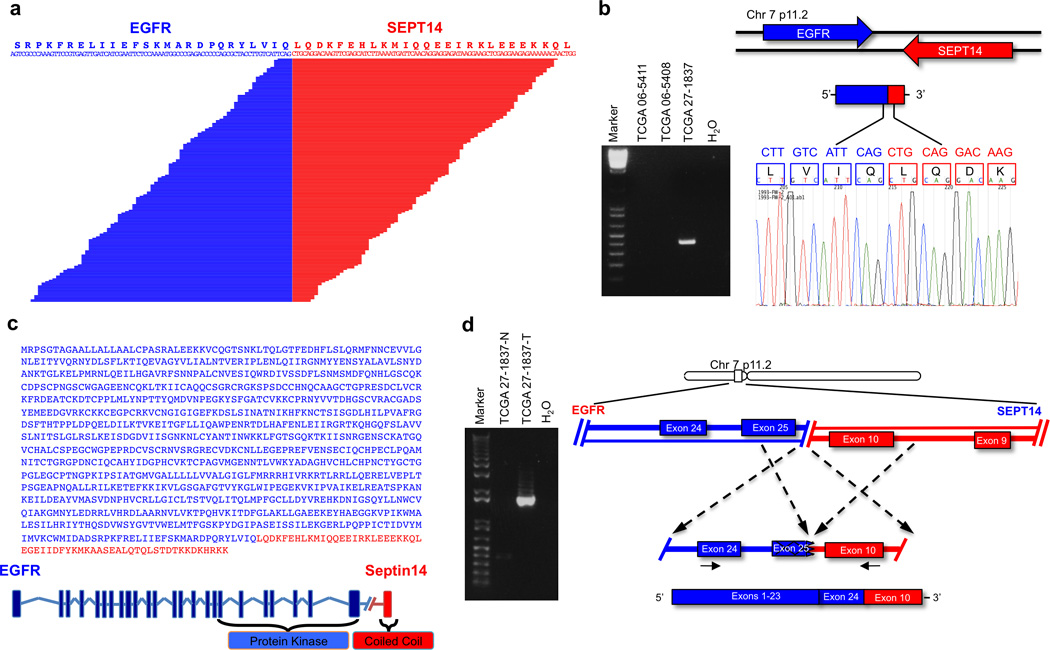
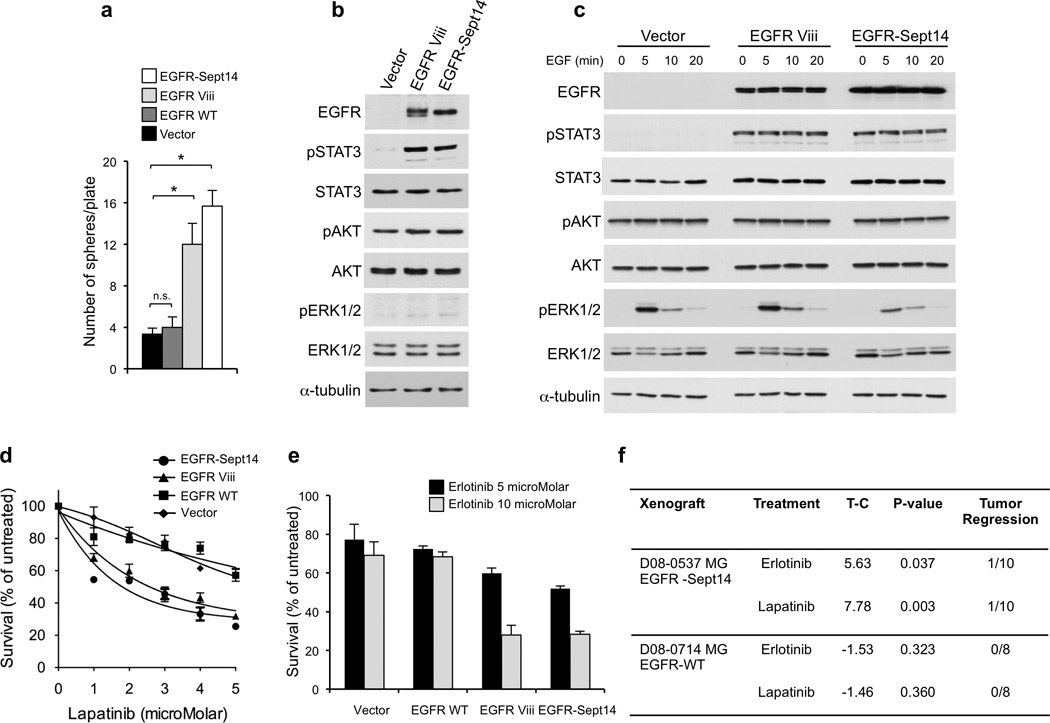
Glioblastoma is one of the most challenging forms of cancer to treat. Here we describe a computational platform that integrates the analysis of copy number variations and somatic mutations and unravels the landscape of in-frame gene fusions in glioblastoma. We found mutations with loss of heterozygosity in LZTR1, encoding an adaptor of CUL3-containing E3 ligase complexes. Mutations and deletions disrupt LZTR1 function, which restrains the self renewal and growth of glioma spheres that retain stem cell features. Loss-of-function mutations in CTNND2 target a neural-specific gene and are associated with the transformation of glioma cells along the very aggressive mesenchymal phenotype. We also report recurrent translocations that fuse the coding sequence of EGFR to several partners, with EGFR-SEPT14 being the most frequent functional gene fusion in human glioblastoma. EGFR-SEPT14 fusions activate STAT3 signaling and confer mitogen independence and sensitivity to EGFR inhibition. These results provide insights into the pathogenesis of glioblastoma and highlight new targets for therapeutic intervention.
Figures
Comment in
-
Genetics: Glioblastoma landscape revealed.Nat Rev Clin Oncol. 2013 Oct;10(10):547. doi: 10.1038/nrclinonc.2013.157. Epub 2013 Aug 20. Nat Rev Clin Oncol. 2013. PMID: 23959268 No abstract available.
-
Genomics informs glioblastoma biology.Nat Genet. 2013 Oct;45(10):1105-7. doi: 10.1038/ng.2775. Nat Genet. 2013. PMID: 24071842
-
Neuro-oncology: Novel molecular targets in treatment of glioblastoma.Nat Rev Neurol. 2013 Nov;9(11):612-3. doi: 10.1038/nrneurol.2013.210. Epub 2013 Oct 15. Nat Rev Neurol. 2013. PMID: 24126628 No abstract available.
References
-
- Stupp R, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Engl. J. Med. 2005;352:987–996. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA127643/CA/NCI NIH HHS/United States
- P30 CA016672/CA/NCI NIH HHS/United States
- NIH 1R01CA164152-01/CA/NCI NIH HHS/United States
- R01 CA085628/CA/NCI NIH HHS/United States
- R01CA131126/CA/NCI NIH HHS/United States
- R01 CA190891/CA/NCI NIH HHS/United States
- R01 CA101644/CA/NCI NIH HHS/United States
- R01CA127643/CA/NCI NIH HHS/United States
- U54 CA121852/CA/NCI NIH HHS/United States
- NIH 1 U54 CA121852-05/CA/NCI NIH HHS/United States
- R01CA085628/CA/NCI NIH HHS/United States
- R01 NS066955/NS/NINDS NIH HHS/United States
- R01 CA164152/CA/NCI NIH HHS/United States
- R01 NS061776/NS/NINDS NIH HHS/United States
- R01NS061776/NS/NINDS NIH HHS/United States
- R01 NS073610/NS/NINDS NIH HHS/United States
- NIH 1 P50 MH094267-01/MH/NIMH NIH HHS/United States
- R01 CA179044/CA/NCI NIH HHS/United States
- R01 CA131126/CA/NCI NIH HHS/United States
- R01CA101644/CA/NCI NIH HHS/United States
- P50 MH094267/MH/NIMH NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
