

Genotype-phenotype correlations in recessive RYR1-related myopathies
- PMID: 23919265
- PMCID: PMC3751094
- DOI: 10.1186/1750-1172-8-117
Genotype-phenotype correlations in recessive RYR1-related myopathies
Abstract
Background: RYR1 mutations are typically associated with core myopathies and are the most common overall cause of congenital myopathy. Dominant mutations are most often associated with central core disease and malignant hyperthermia, and genotype-phenotype patterns have emerged from the study of these mutations that have contributed to the understanding of disease pathogenesis. The recent availability of genetic testing for the entire RYR1 coding sequence has led to a dramatic expansion in the identification of recessive mutations in core myopathies and other congenital myopathies. To date, no clear patterns have been identified in these recessive mutations, though no systematic examination has yet been performed.
Methods: In this study, we investigated genotype-phenotype correlations in a large combined cohort of unpublished (n = 14) and previously reported (n = 92) recessive RYR1 cases.
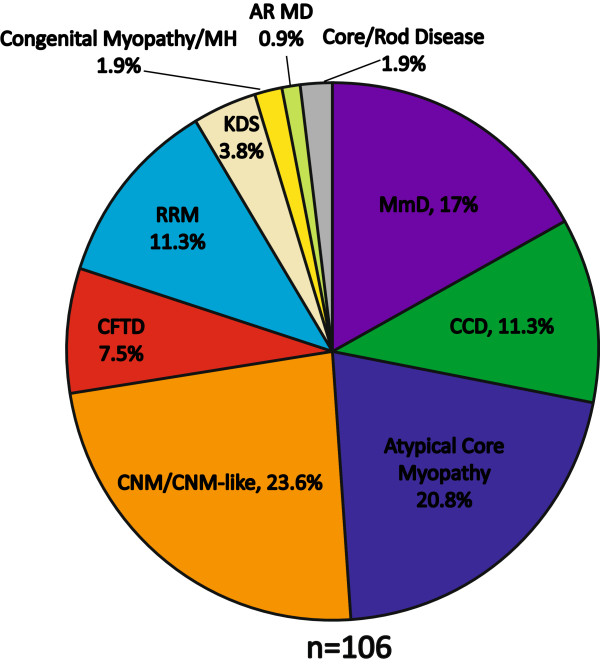
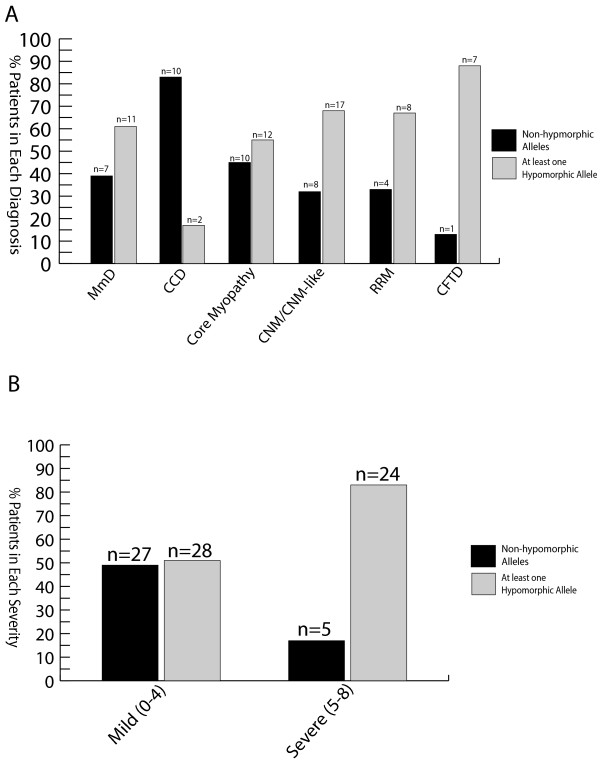
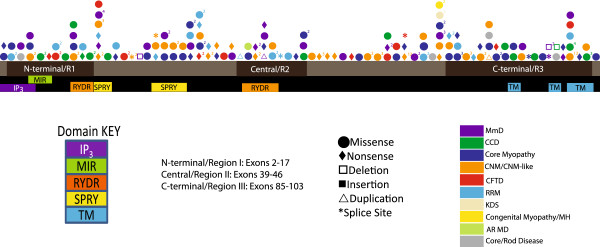
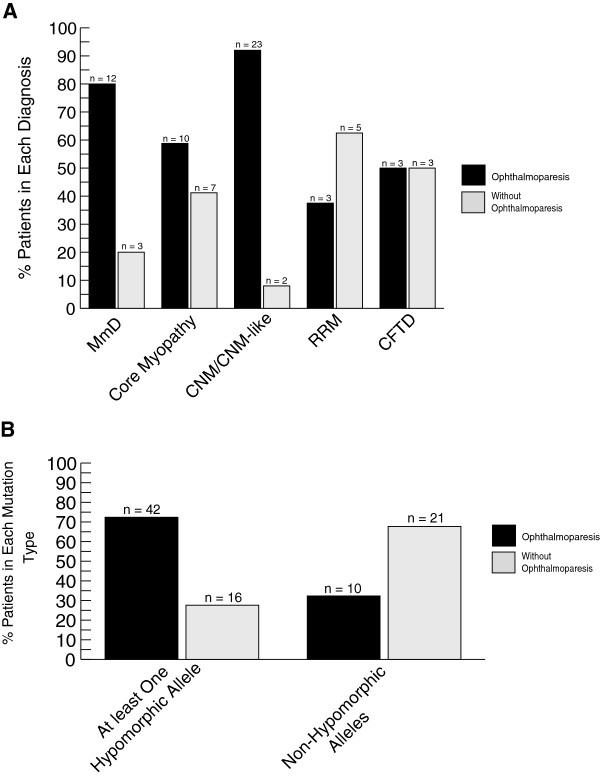
Results: Overall examination of this cohort revealed nearly 50% of cases to be non-core myopathy related. Our most significant finding was that hypomorphic mutations (mutations expected to diminish RyR1 expression) were enriched in patients with severe clinical phenotypes. We also determined that hypomorphic mutations were more likely to be encountered in non-central core myopathies. With analysis of the location of non-hypomorphic mutations, we found that missense mutations were generally enriched in the MH/CCD hotspots and specifically enriched in the selectivity filter of the channel pore.
Conclusions: These results support a hypothesis that loss of protein function is a key predictive disease parameter. In addition, they suggest that decreased RyR1 expression may dictate non-core related pathology though, data on protein expression was limited and should be confirmed in a larger cohort. Lastly, the results implicate abnormal ion conductance through the channel pore in the pathogenesis in recessive core myopathies. Overall, our findings represent a comprehensive analysis of genotype-phenotype associations in recessive RYR1-myopathies.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
