

Sources of bias in measures of allele-specific expression derived from RNA-sequence data aligned to a single reference genome
- PMID: 23919664
- PMCID: PMC3751238
- DOI: 10.1186/1471-2164-14-536
Sources of bias in measures of allele-specific expression derived from RNA-sequence data aligned to a single reference genome
Abstract
Background: RNA-seq can be used to measure allele-specific expression (ASE) by assigning sequence reads to individual alleles; however, relative ASE is systematically biased when sequence reads are aligned to a single reference genome. Aligning sequence reads to both parental genomes can eliminate this bias, but this approach is not always practical, especially for non-model organisms. To improve accuracy of ASE measured using a single reference genome, we identified properties of differentiating sites responsible for biased measures of relative ASE.
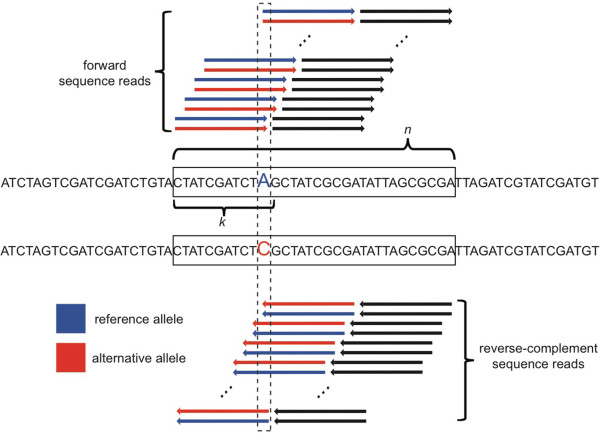
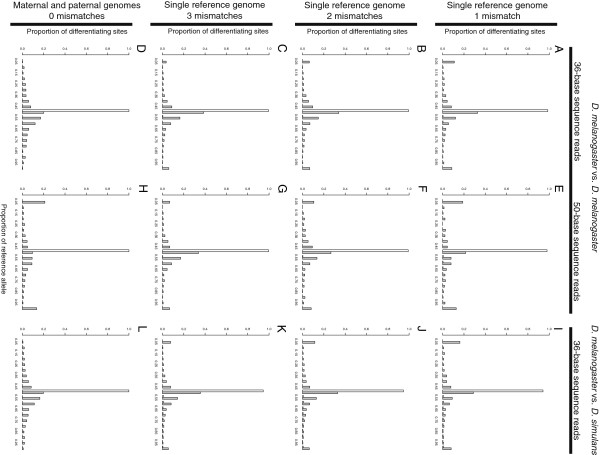
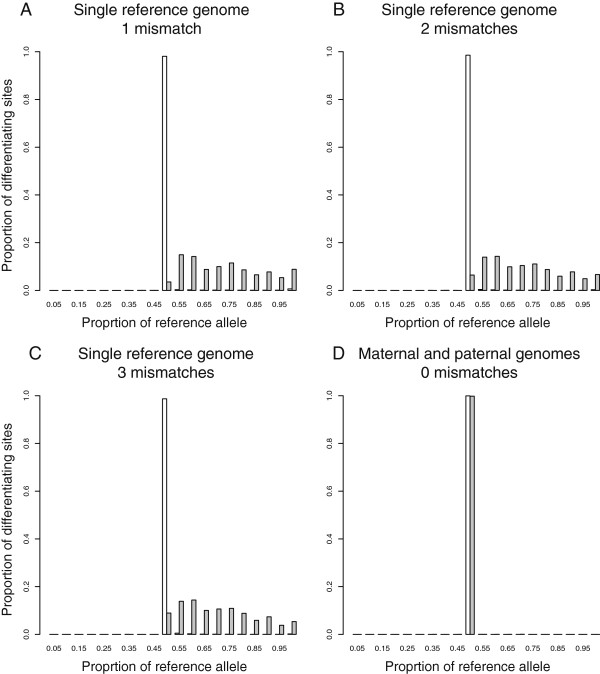
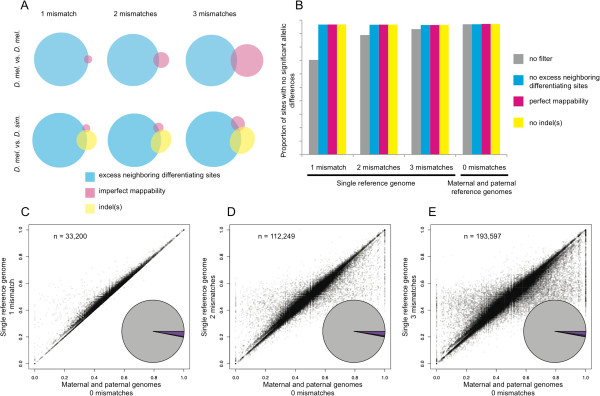
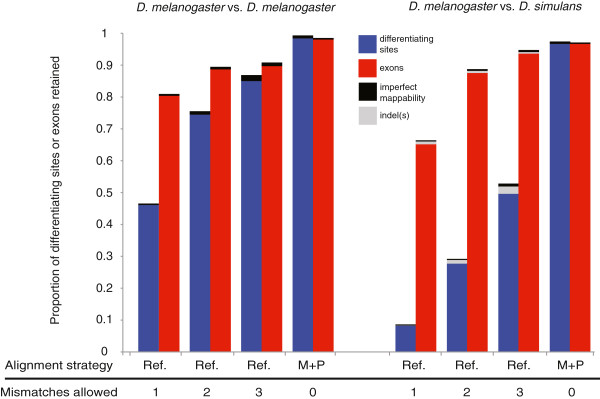
Results: We found that clusters of differentiating sites prevented sequence reads from an alternate allele from aligning to the reference genome, causing a bias in relative ASE favoring the reference allele. This bias increased with greater sequence divergence between alleles. Increasing the number of mismatches allowed when aligning sequence reads to the reference genome and restricting analysis to genomic regions with fewer differentiating sites than the number of mismatches allowed almost completely eliminated this systematic bias. Accuracy of allelic abundance was increased further by excluding differentiating sites within sequence reads that could not be aligned uniquely within the genome (imperfect mappability) and reads that overlapped one or more insertions or deletions (indels) between alleles.
Conclusions: After aligning sequence reads to a single reference genome, excluding differentiating sites with at least as many neighboring differentiating sites as the number of mismatches allowed, imperfect mappability, and/or an indel(s) nearby resulted in measures of allelic abundance comparable to those derived from aligning sequence reads to both parental genomes.
Figures

References
-
- Brawand D, Soumillon M, Necsulea A, Julien P, Csárdi G, Harrigan P, Weier M, Liechti A, Aximu-Petri A, Kircher M, Albert FW, Zeller U, Khaitovich P, Grützner F, Bergmann S, Nielsen R, Pääbo S, Kaessmann H. The evolution of gene expression levels in mammalian organs. Nature. 2011;478:343–348. doi: 10.1038/nature10532. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
