

The solution structure of heparan sulfate differs from that of heparin: implications for function
- PMID: 23921391
- PMCID: PMC3784691
- DOI: 10.1074/jbc.M113.492223
The solution structure of heparan sulfate differs from that of heparin: implications for function
Abstract
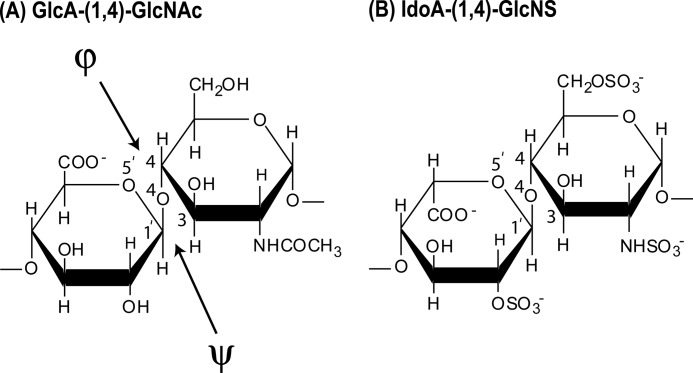
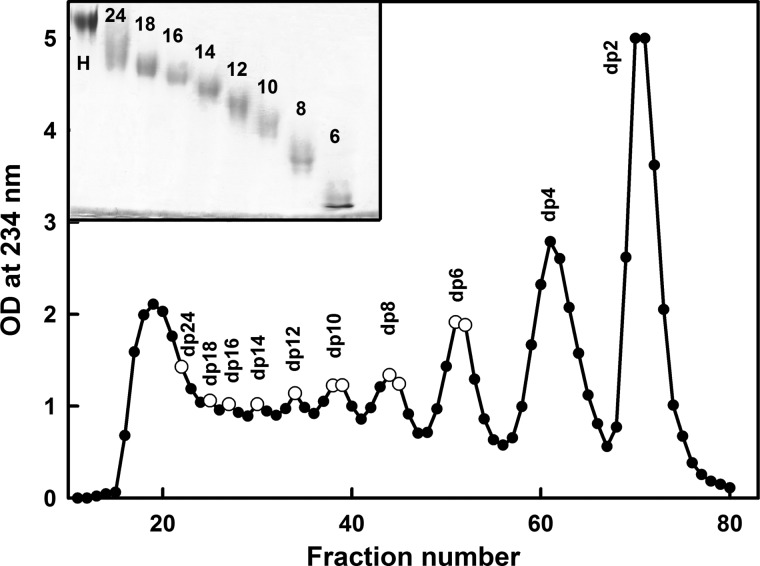
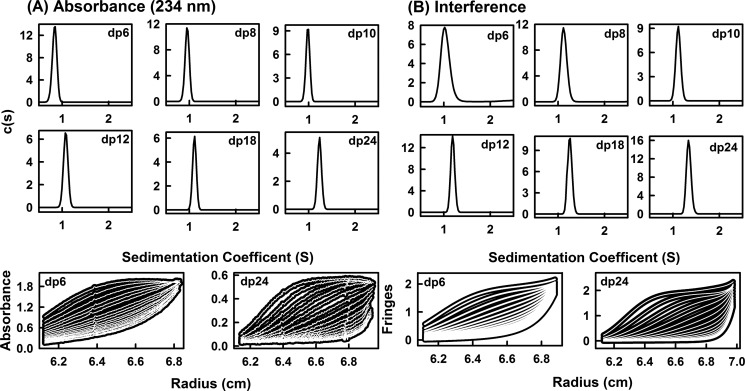
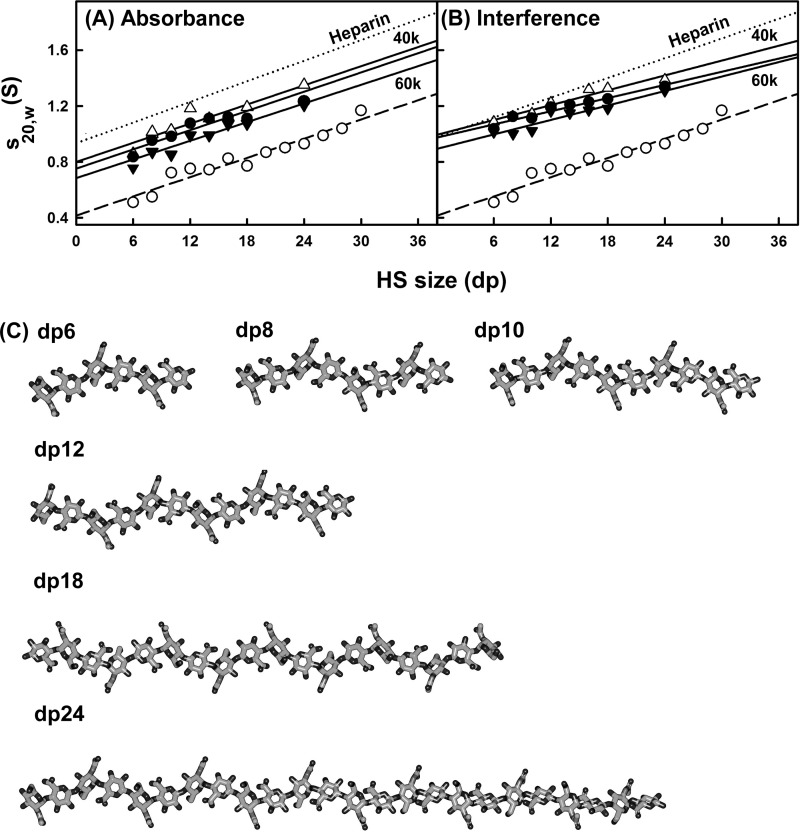
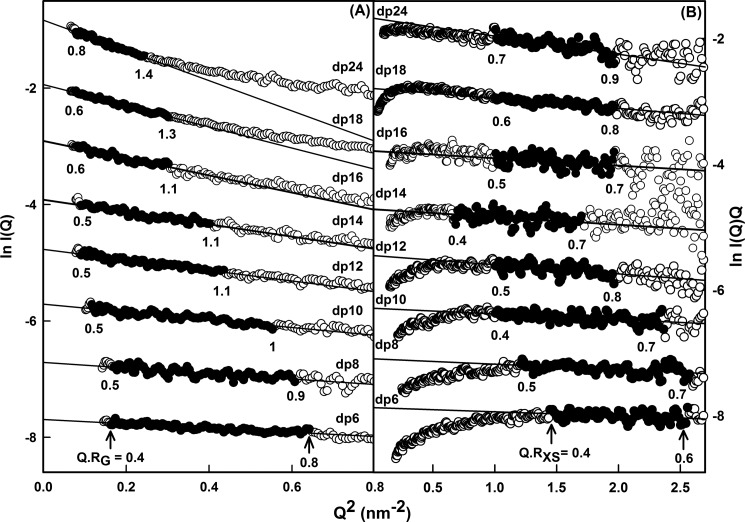
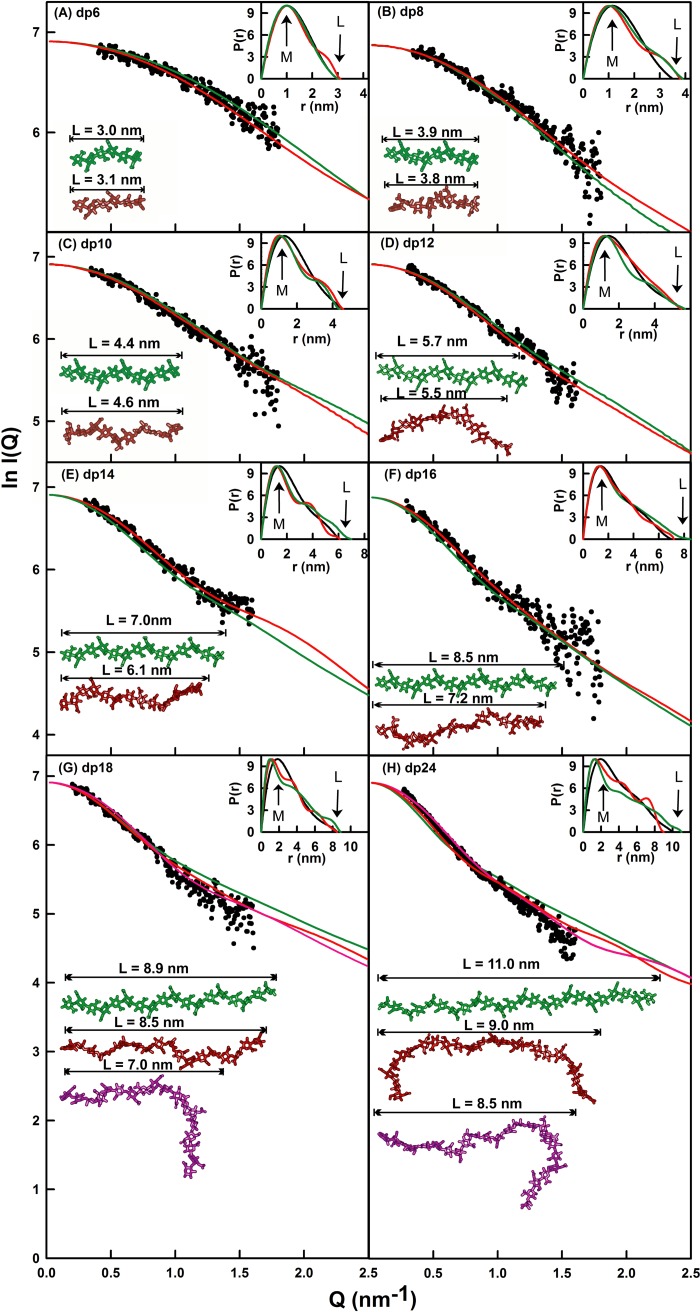
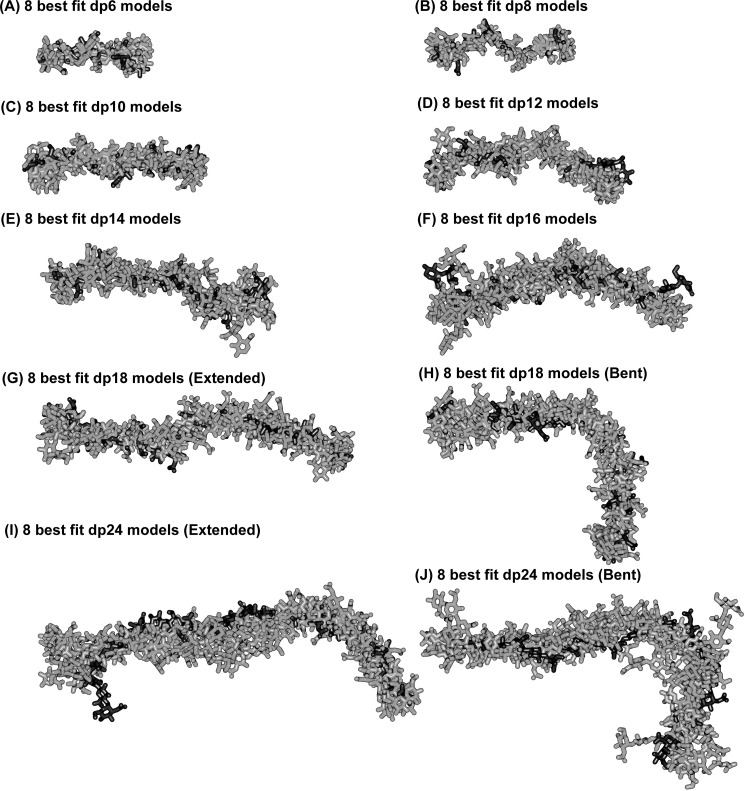
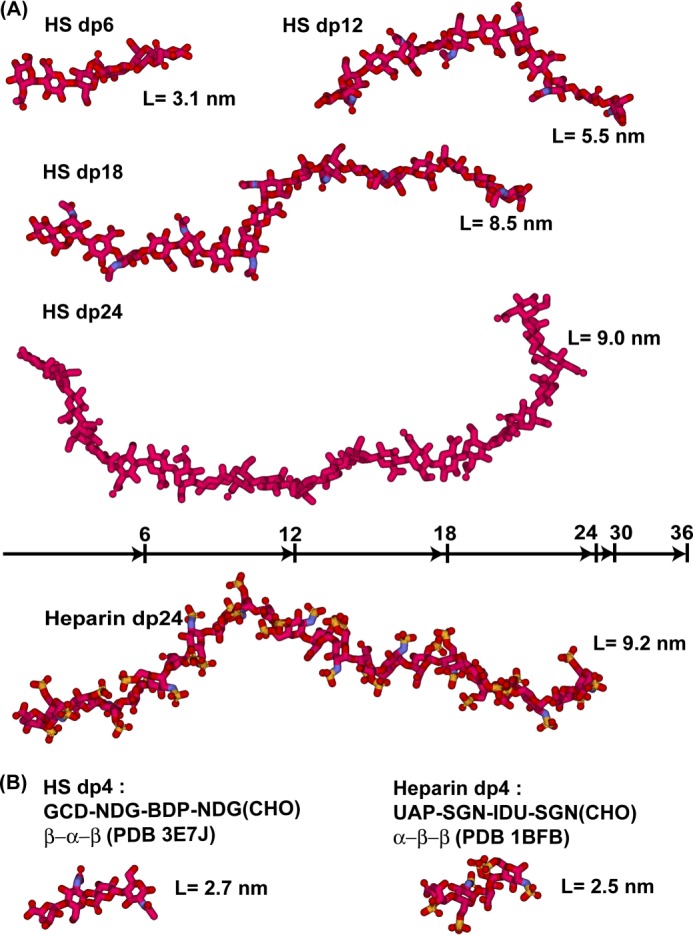
The highly sulfated polysaccharides heparin and heparan sulfate (HS) play key roles in the regulation of physiological and pathophysiological processes. Despite its importance, no molecular structures of free HS have been reported up to now. By combining analytical ultracentrifugation, small angle x-ray scattering, and constrained scattering modeling recently used for heparin, we have analyzed the solution structures for eight purified HS fragments dp6-dp24 corresponding to the predominantly unsulfated GlcA-GlcNAc domains of heparan sulfate. Unlike heparin, the sedimentation coefficient s20,w of HS dp6-dp24 showed a small rotor speed dependence, where similar s20,w values of 0.82-1.26 S (absorbance optics) and 1.05-1.34 S (interference optics) were determined. The corresponding x-ray scattering measurements of HS dp6-dp24 gave radii of gyration RG values from 1.03 to 2.82 nm, cross-sectional radii of gyration RXS values from 0.31 to 0.65 nm, and maximum lengths L from 3.0 to 10.0 nm. These data showed that HS has a longer and more bent structure than heparin. Constrained scattering modeling starting from 5,000 to 12,000 conformationally randomized HS structures gave best fit dp6-dp24 molecular structures that were longer and more bent than their equivalents in heparin. Alternative fits were obtained for HS dp18 and dp24, indicating their higher bending and flexibility. We conclude that HS displays bent conformations that are significantly distinct from that for heparin. The difference is attributed to the different predominant monosaccharide sequence and reduced sulfation of HS, indicating that HS may interact differently with proteins compared with heparin.
Keywords: Analytical Ultracentrifugation; Heparan Sulfate; Heparin; Heparin-binding Protein; Molecular Modeling; X-ray Scattering.
Figures

References
-
- Gallagher J. T., Turnbull J. E., Lyon M. (1992) Heparan sulphate proteoglycans. Molecular organisation of membrane-associated species and an approach to polysaccharide sequence analysis. Adv. Exp. Med. Biol. 313, 49–57 - PubMed
-
- Bernfield M., Kokenyesi R., Kato M., Hinkes M. T., Spring J., Gallo R. L., Lose E. J. (1992) Biology of the syndecans. A family of transmembrane heparan sulfate proteoglycans. Annu. Rev. Cell Biol. 8, 365–393 - PubMed
-
- Conrad H. E. (1998) Heparin-binding Proteins, Academic Press, San Diego
-
- Sasisekharan R., Venkataraman G. (2000) Heparin and heparan sulfate. Biosynthesis, structure and function. Curr. Opin. Chem. Biol. 4, 626–631 - PubMed
-
- Perrimon N., Bernfield M. (2000) Specificities of heparan sulphate proteoglycans in developmental processes. Nature 404, 725–728 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
