

Detecting Alu insertions from high-throughput sequencing data
- PMID: 23921633
- PMCID: PMC3783187
- DOI: 10.1093/nar/gkt612
Detecting Alu insertions from high-throughput sequencing data
Abstract
High-throughput sequencing technologies have allowed for the cataloguing of variation in personal human genomes. In this manuscript, we present alu-detect, a tool that combines read-pair and split-read information to detect novel Alus and their precise breakpoints directly from either whole-genome or whole-exome sequencing data while also identifying insertions directly in the vicinity of existing Alus. To set the parameters of our method, we use simulation of a faux reference, which allows us to compute the precision and recall of various parameter settings using real sequencing data. Applying our method to 100 bp paired Illumina data from seven individuals, including two trios, we detected on average 1519 novel Alus per sample. Based on the faux-reference simulation, we estimate that our method has 97% precision and 85% recall. We identify 808 novel Alus not previously described in other studies. We also demonstrate the use of alu-detect to study the local sequence and global location preferences for novel Alu insertions.
Figures
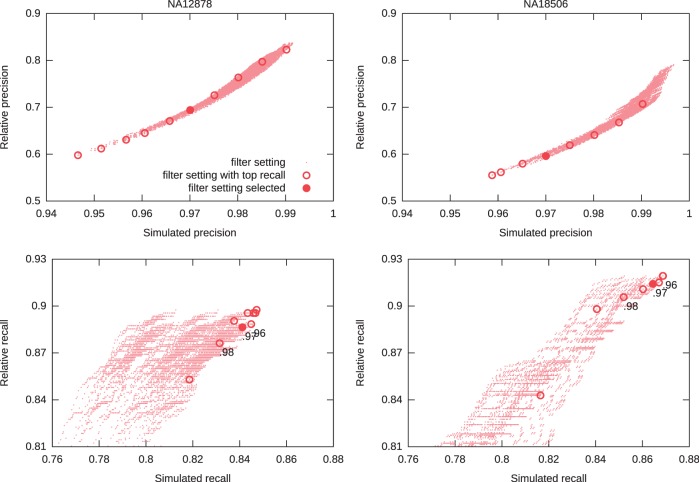
 ) versus Simulated Precision (
) versus Simulated Precision ( ). Bottom: Relative Recall (
). Bottom: Relative Recall ( ) versus Simulated Recall (
) versus Simulated Recall ( ). Each dot represents a filter setting. We highlight, for x between 0.920 and 0.990 in steps of 0.005, the filter with
). Each dot represents a filter setting. We highlight, for x between 0.920 and 0.990 in steps of 0.005, the filter with  and maximum simulated recall
and maximum simulated recall  . NA12878 is an individual of European ancestry, whereas NA18506 is an individual of Yoruban ancestry.
. NA12878 is an individual of European ancestry, whereas NA18506 is an individual of Yoruban ancestry.

 is highlighted). We also show data points corresponding to the results of (6) and (7). Bottom: Intersections of calls on each sample between studies (area-proportional). Next to the study identifier: calls made and relative precision (fraction of the calls also present in dbRIP or Stewart et al.). The numbers in the diagram do not always add up because interval intersection is non-transitive.
is highlighted). We also show data points corresponding to the results of (6) and (7). Bottom: Intersections of calls on each sample between studies (area-proportional). Next to the study identifier: calls made and relative precision (fraction of the calls also present in dbRIP or Stewart et al.). The numbers in the diagram do not always add up because interval intersection is non-transitive.References
-
- Dalca AV, Brudno M. Genome variation discovery with high-throughput sequencing data. Brief. Bioinformatics. 2010;11:3–14. - PubMed
-
- Medvedev P, Stanciu M, Brudno M. Computational methods for discovering structural variation with next-generation sequencing. Nat. Methods. 2009;6(11 Suppl.):S13–S20. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
