

Epigenetic and genetic influences on DNA methylation variation in maize populations
- PMID: 23922207
- PMCID: PMC3784580
- DOI: 10.1105/tpc.113.114793
Epigenetic and genetic influences on DNA methylation variation in maize populations
Abstract
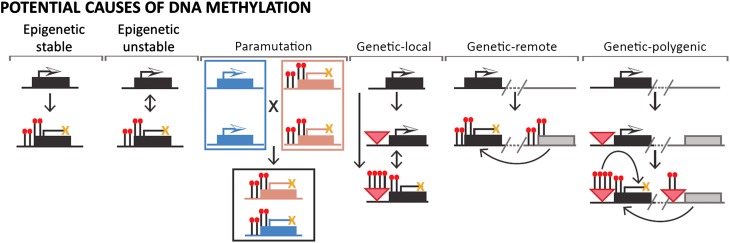
DNA methylation is a chromatin modification that is frequently associated with epigenetic regulation in plants and mammals. However, genetic changes such as transposon insertions can also lead to changes in DNA methylation. Genome-wide profiles of DNA methylation for 20 maize (Zea mays) inbred lines were used to discover differentially methylated regions (DMRs). The methylation level for each of these DMRs was also assayed in 31 additional maize or teosinte genotypes, resulting in the discovery of 1966 common DMRs and 1754 rare DMRs. Analysis of recombinant inbred lines provides evidence that the majority of DMRs are heritable. A local association scan found that nearly half of the DMRs with common variation are significantly associated with single nucleotide polymorphisms found within or near the DMR. Many of the DMRs that are significantly associated with local genetic variation are found near transposable elements that may contribute to the variation in DNA methylation. Analysis of gene expression in the same samples used for DNA methylation profiling identified over 300 genes with expression patterns that are significantly associated with DNA methylation variation. Collectively, our results suggest that DNA methylation variation is influenced by genetic and epigenetic changes that are often stably inherited and can influence the expression of nearby genes.
Figures






References
-
- Becker C., Hagmann J., Müller J., Koenig D., Stegle O., Borgwardt K., Weigel D. (2011). Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 480: 245–249 - PubMed
-
- Bender J., Fink G.R. (1995). Epigenetic control of an endogenous gene family is revealed by a novel blue fluorescent mutant of Arabidopsis. Cell 83: 725–734 - PubMed
-
- Bird A. (2007). Perceptions of epigenetics. Nature 447: 396–398 - PubMed
-
- Buckler E.S., Gaut B.S., McMullen M.D. (2006). Molecular and functional diversity of maize. Curr. Opin. Plant Biol. 9: 172–176 - PubMed
Publication types
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
