Efficient exploration of the space of reconciled gene trees
- PMID: 23925510
- PMCID: PMC3797637
- DOI: 10.1093/sysbio/syt054
Efficient exploration of the space of reconciled gene trees
Abstract
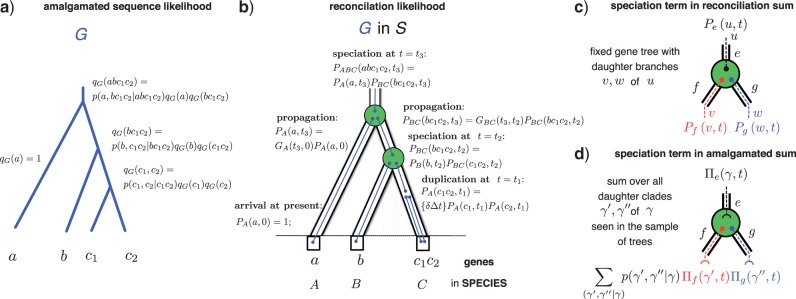
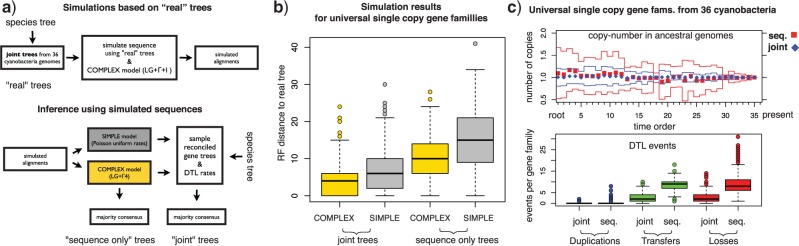
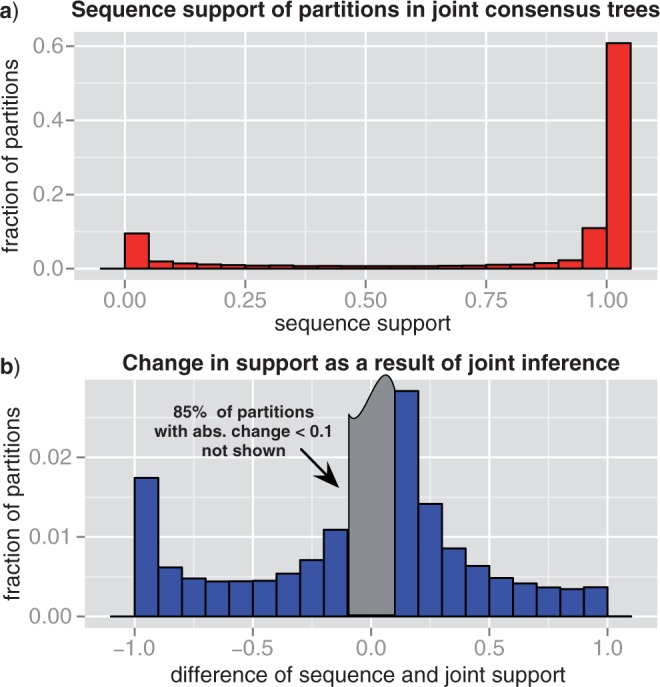
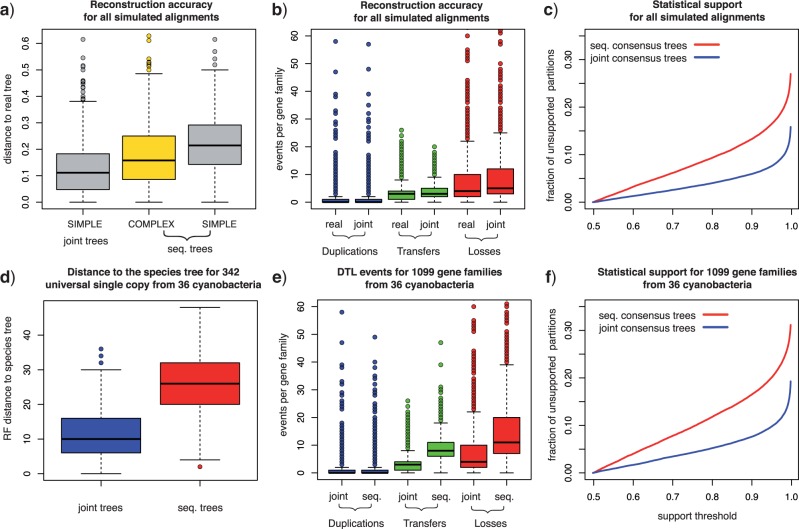
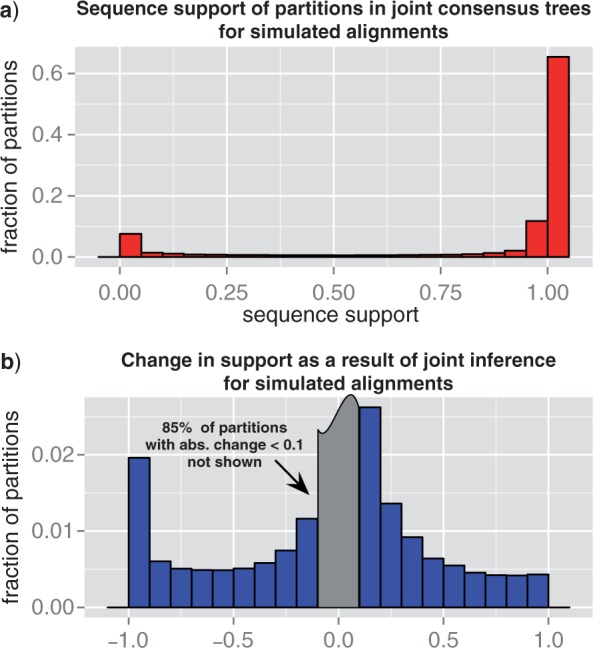
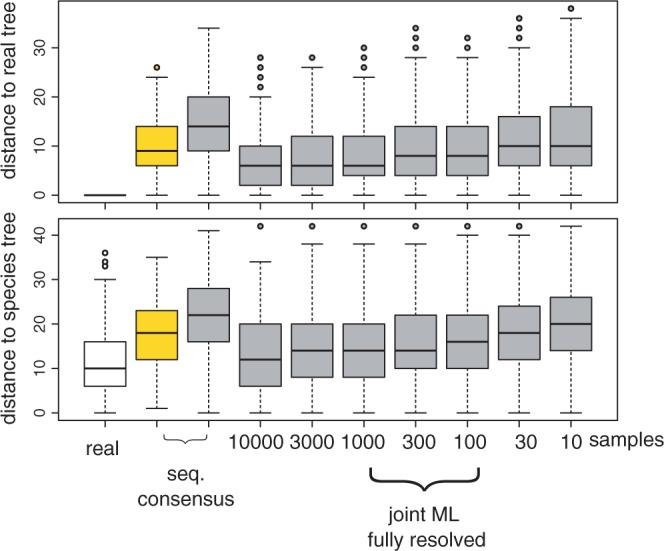
Gene trees record the combination of gene-level events, such as duplication, transfer and loss (DTL), and species-level events, such as speciation and extinction. Gene tree-species tree reconciliation methods model these processes by drawing gene trees into the species tree using a series of gene and species-level events. The reconstruction of gene trees based on sequence alone almost always involves choosing between statistically equivalent or weakly distinguishable relationships that could be much better resolved based on a putative species tree. To exploit this potential for accurate reconstruction of gene trees, the space of reconciled gene trees must be explored according to a joint model of sequence evolution and gene tree-species tree reconciliation. Here we present amalgamated likelihood estimation (ALE), a probabilistic approach to exhaustively explore all reconciled gene trees that can be amalgamated as a combination of clades observed in a sample of gene trees. We implement the ALE approach in the context of a reconciliation model (Szöllősi et al. 2013), which allows for the DTL of genes. We use ALE to efficiently approximate the sum of the joint likelihood over amalgamations and to find the reconciled gene tree that maximizes the joint likelihood among all such trees. We demonstrate using simulations that gene trees reconstructed using the joint likelihood are substantially more accurate than those reconstructed using sequence alone. Using realistic gene tree topologies, branch lengths, and alignment sizes, we demonstrate that ALE produces more accurate gene trees even if the model of sequence evolution is greatly simplified. Finally, examining 1099 gene families from 36 cyanobacterial genomes we find that joint likelihood-based inference results in a striking reduction in apparent phylogenetic discord, with respectively. 24%, 59%, and 46% reductions in the mean numbers of duplications, transfers, and losses per gene family. The open source implementation of ALE is available from https://github.com/ssolo/ALE.git.
Figures