

The population genomics of repeated evolution in the blind cavefish Astyanax mexicanus
- PMID: 23927992
- PMCID: PMC3808867
- DOI: 10.1093/molbev/mst136
The population genomics of repeated evolution in the blind cavefish Astyanax mexicanus
Abstract
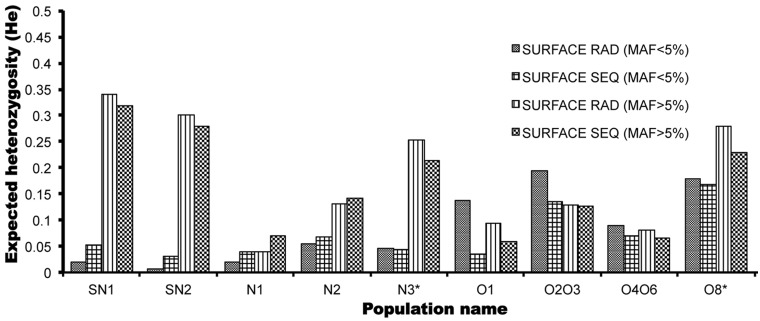
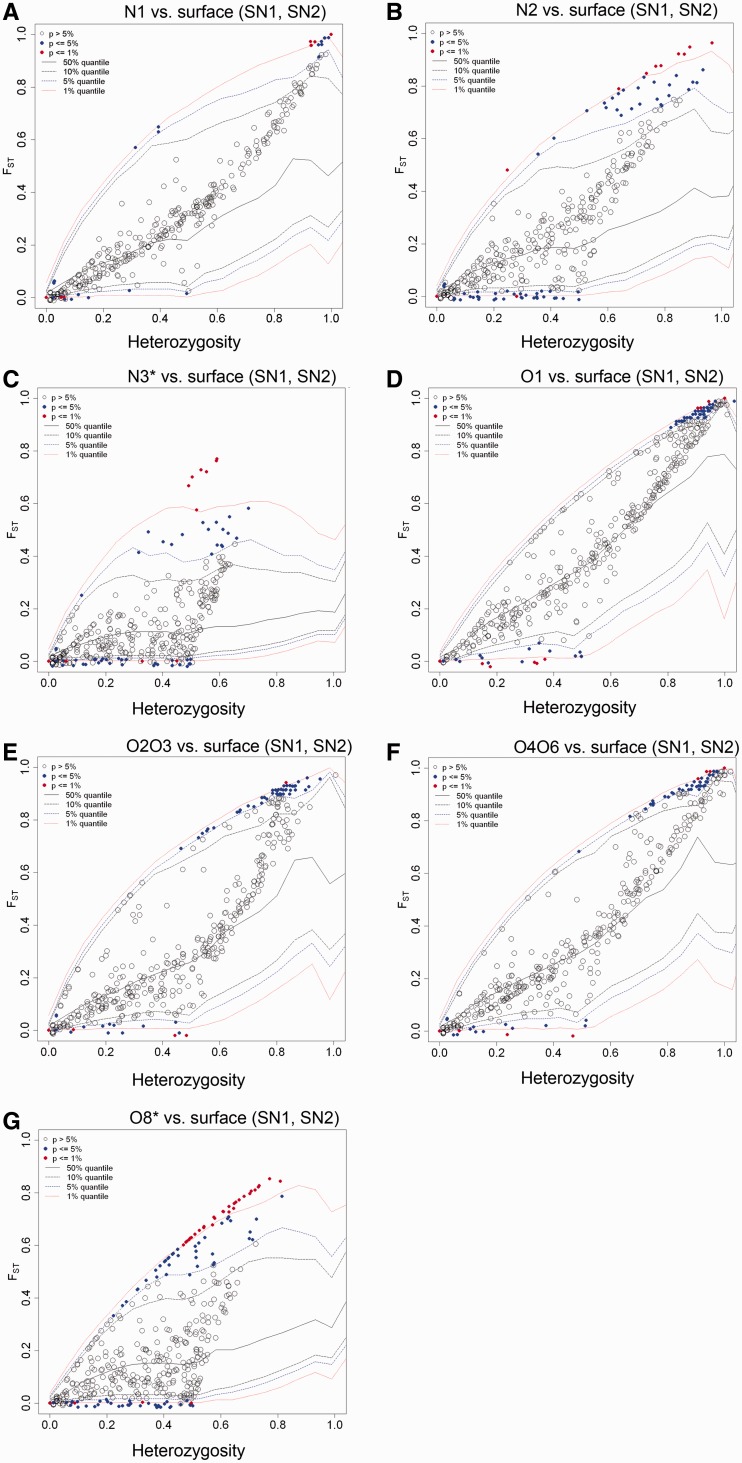
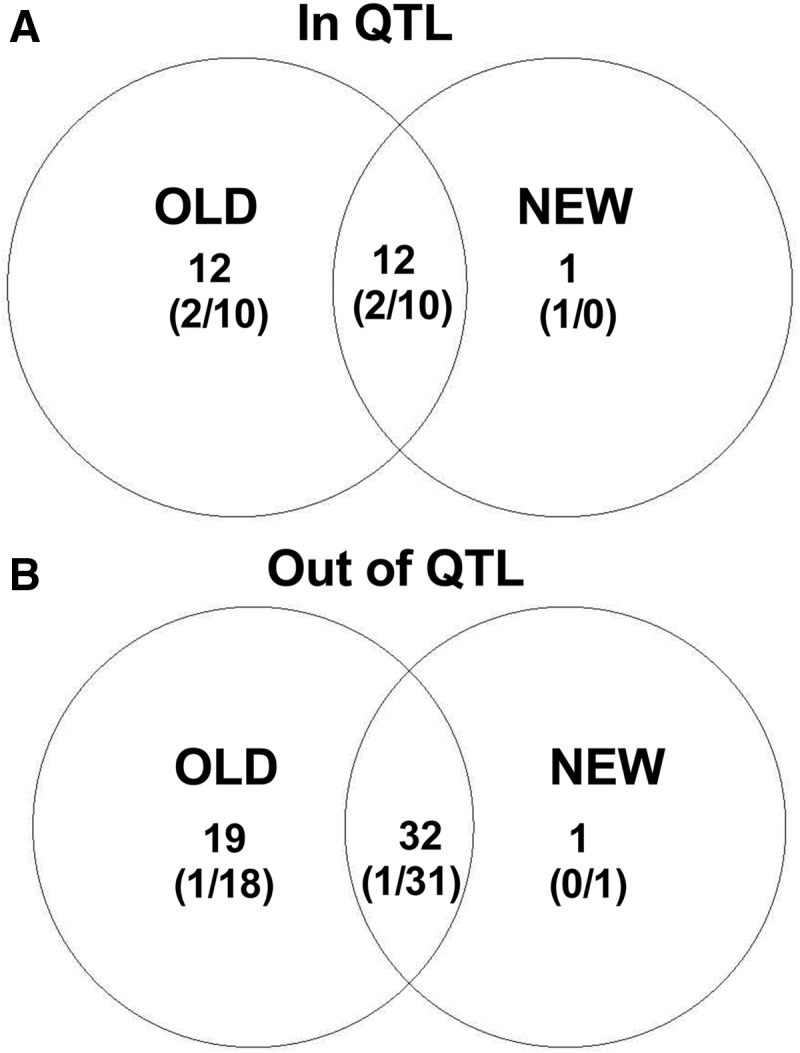
Distinct populations of Astyanax mexicanus cavefish offer striking examples of repeatable convergence or parallelism in their independent evolutions from surface to cave phenotypes. However, the extent to which the repeatability of evolution occurred at the genetic level remains poorly understood. To address this, we first characterized the genetic diversity of 518 single-nucleotide polymorphisms (SNPs), obtained through RAD tag sequencing and distributed throughout the genome, in seven cave and three groups of surface populations. The cave populations represented two distinct lineages (old and new). Thirty-one SNPs were significantly differentiated between surface and old cave populations, two SNPs were differentiated between surface and new cave populations, and 44 SNPs were significantly differentiated in both old and new cave populations. In addition, we determined whether these SNPs map to the same locations of previously described quantitative trait loci (QTL) between surface and cave populations. A total of 25 differentiated SNPs co-map with several QTL, such as one containing a fibroblast growth factor gene (Fgf8) involved in eye development and lens size. Further, the identity of many SNPs that co-mapped with QTL was the same in independently derived cave populations. These conclusions were further confirmed by haplotype analyses of SNPs within QTL regions. Our findings indicate that the repeatability of evolution at the genetic level is substantial, suggesting that ancestral standing genetic variation significantly contributed to the population genetic variability used in adaptation to the cave environment.
Keywords: QTL; RAD tag sequencing; SNP; convergence; parallelism; standing variation.
Figures


References
-
- Arendt J, Reznick D. Convergence and parallelism reconsidered: what have we learned about the genetics of adaptation? Trends Ecol Evol. 2008;23:26–32. - PubMed
-
- Barrett RD, Rogers SM, Schluter D. Environment specific pleiotropy facilitates divergence at the Ectodysplasin locus in threespine stickleback. Evolution. 2009;63:2831–2837. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
