

Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure
- PMID: 23931755
- PMCID: PMC3779647
- DOI: 10.1016/j.cmet.2013.07.002
Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure
Abstract
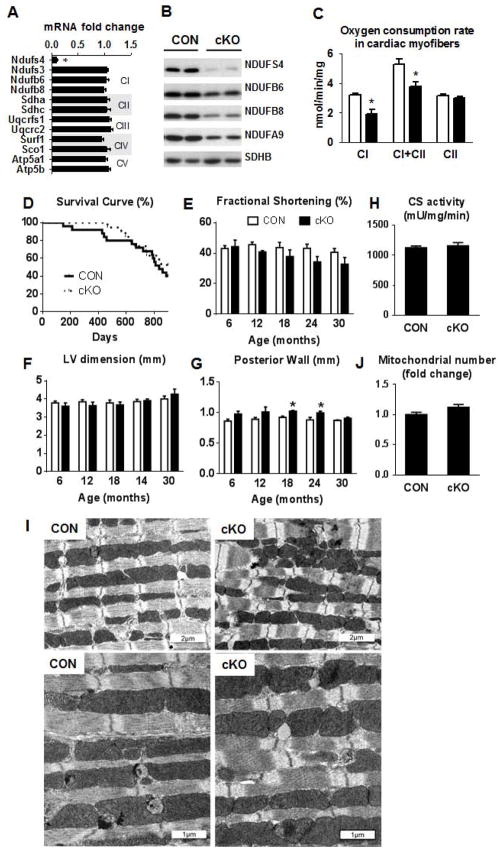
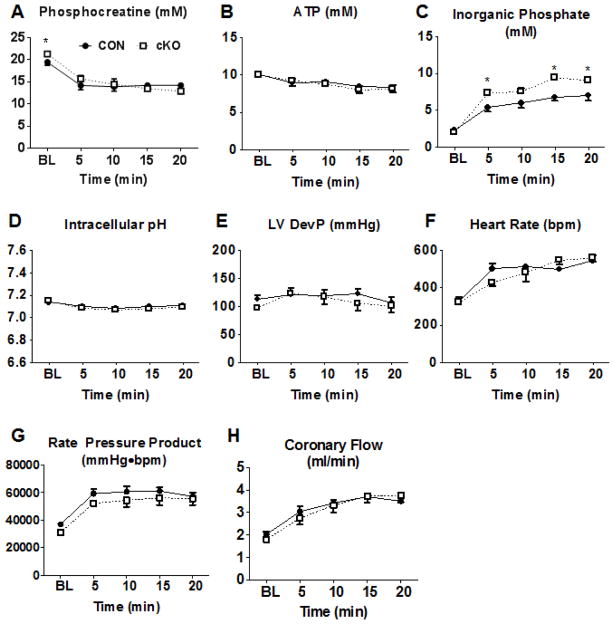
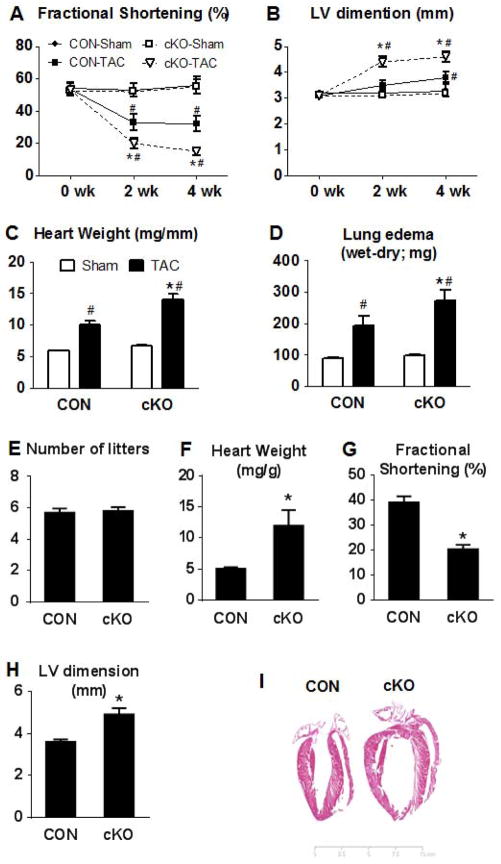
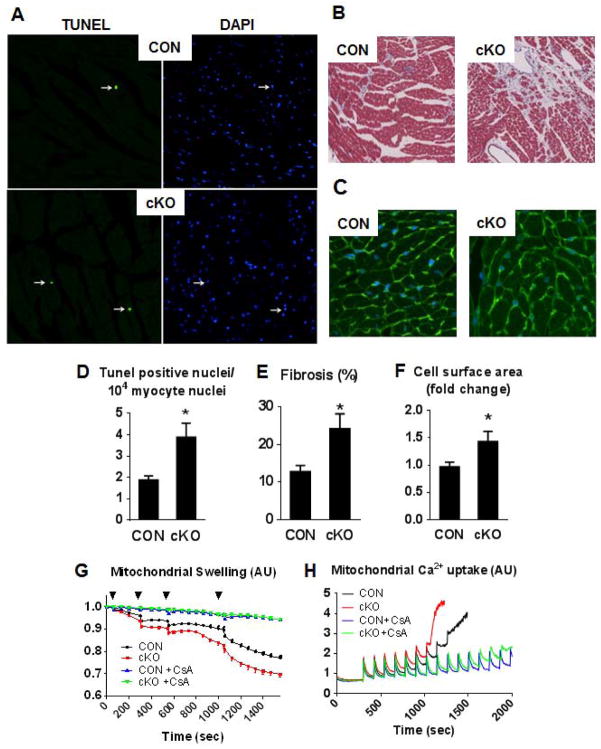
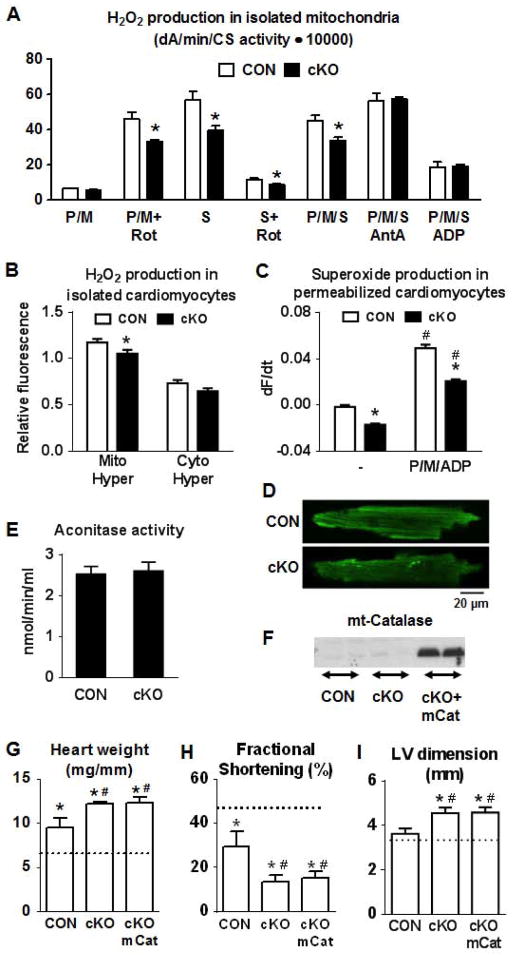
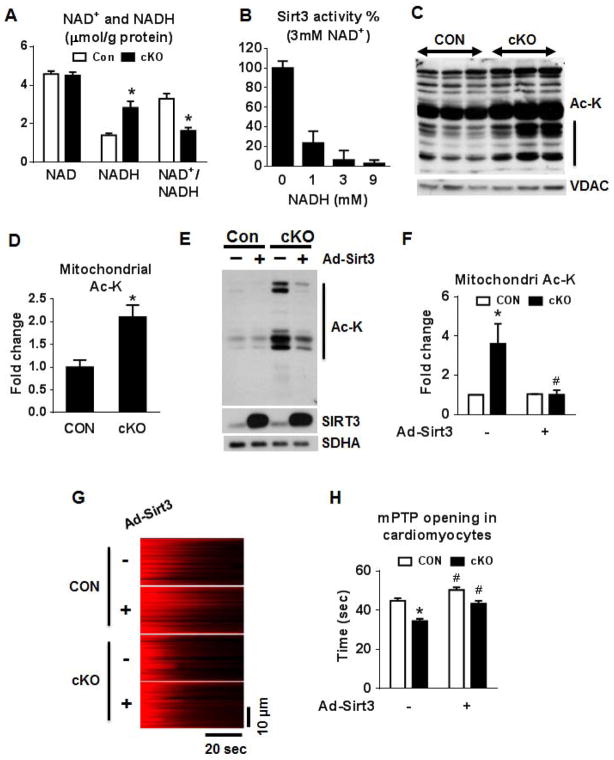
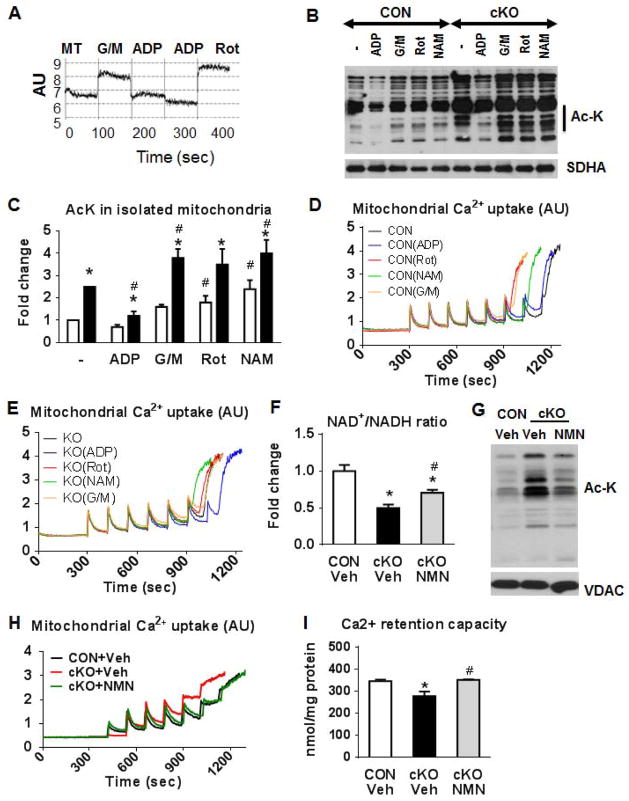
Mitochondrial respiratory dysfunction is linked to the pathogenesis of multiple diseases, including heart failure, but the specific mechanisms for this link remain largely elusive. We modeled the impairment of mitochondrial respiration by the inactivation of the Ndufs4 gene, a protein critical for complex I assembly, in the mouse heart (cKO). Although complex I-supported respiration decreased by >40%, the cKO mice maintained normal cardiac function in vivo and high-energy phosphate content in isolated perfused hearts. However, the cKO mice developed accelerated heart failure after pressure overload or repeated pregnancy. Decreased NAD(+)/NADH ratio by complex I deficiency inhibited Sirt3 activity, leading to an increase in protein acetylation and sensitization of the permeability transition in mitochondria (mPTP). NAD(+) precursor supplementation to cKO mice partially normalized the NAD(+)/NADH ratio, protein acetylation, and mPTP sensitivity. These findings describe a mechanism connecting mitochondrial dysfunction to the susceptibility to diseases and propose a potential therapeutic target.
Copyright © 2013 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Avalos JL, Bever KM, Wolberger C. Mechanism of sirtuin inhibition by nicotinamide: altering the NAD(+) cosubstrate specificity of a Sir2 enzyme. Molecular cell. 2005;17:855–868. - PubMed
-
- Boehm EA, Jones BE, Radda GK, Veech RL, Clarke K. Increased uncoupling proteins and decreased efficiency in palmitate-perfused hyperthyroid rat heart. American journal of physiology. 2001;280:H977–983. - PubMed
-
- DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. The New England journal of medicine. 2003;348:2656–2668. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
