

Congenital myopathy is caused by mutation of HACD1
- PMID: 23933735
- PMCID: PMC3842179
- DOI: 10.1093/hmg/ddt380
Congenital myopathy is caused by mutation of HACD1
Abstract
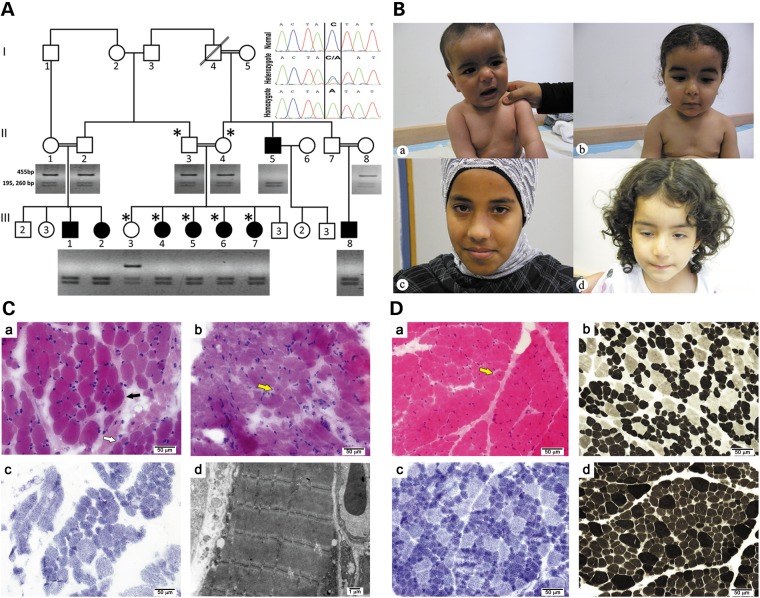
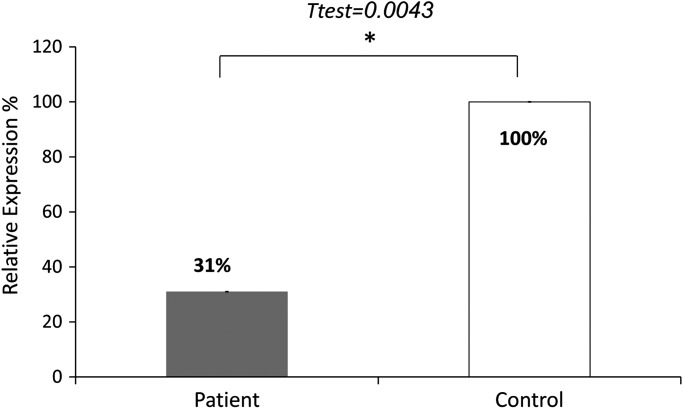
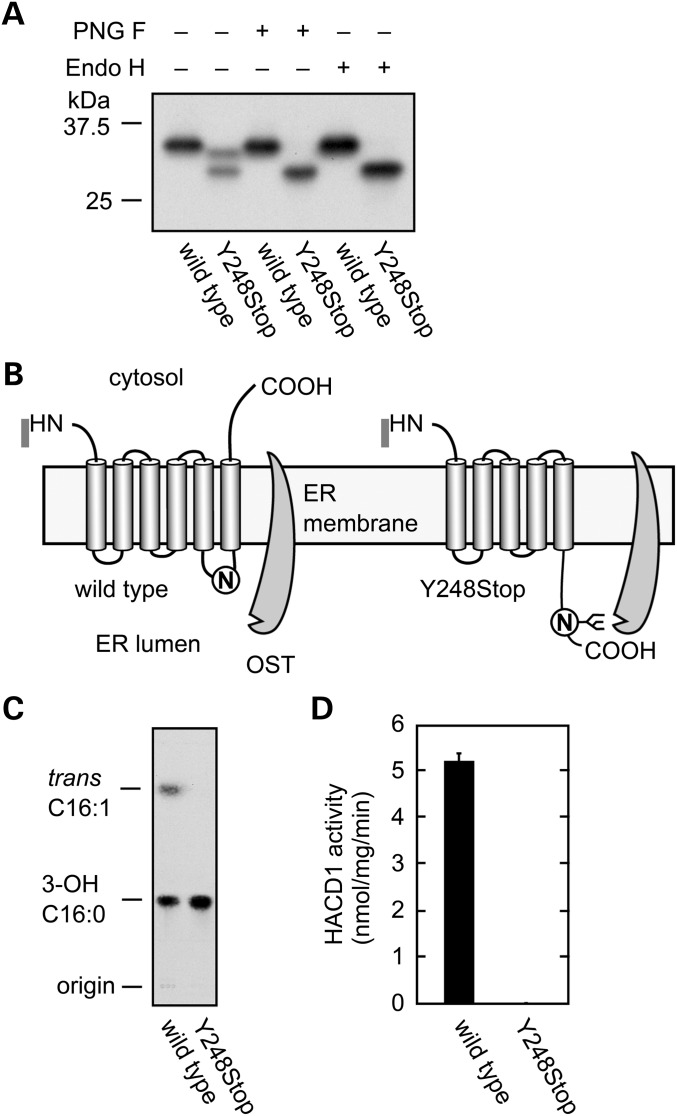
Congenital myopathies are heterogeneous inherited diseases of muscle characterized by a range of distinctive histologic abnormalities. We have studied a consanguineous family with congenital myopathy. Genome-wide linkage analysis and whole-exome sequencing identified a homozygous non-sense mutation in 3-hydroxyacyl-CoA dehydratase 1 (HACD1) in affected individuals. The mutation results in non-sense mediated decay of the HACD1 mRNA to 31% of control levels in patient muscle and completely abrogates the enzymatic activity of dehydration of 3-hydroxyacyl-CoA, the third step in the elongation of very long-chain fatty acids (VLCFAs). We describe clinical findings correlated with a deleterious mutation in a gene not previously known to be associated with congenital myopathy in humans. We suggest that the mutation in the HACD1 gene causes a reduction in the synthesis of VLCFAs, which are components of membrane lipids and participants in physiological processes, leading to congenital myopathy. These data indicate that HACD1 is necessary for muscle function.
Figures
References
-
- Nance J.R., Dowling J.J., Gibbs E.M., Bönnemann C.G. Congenital myopathies: an update. Curr. Neurol. Neurosci. Rep. 2011;12:165–174. doi:10.1007/s11910-012-0255-x. - DOI - PMC - PubMed
-
- Kaplan J.C. The 2012 version of the gene table of monogenic neuromuscular disorders. Neuromuscul. Disord. 2011;21:833–861. doi:10.1016/j.nmd.2011.10.008. - DOI - PubMed
-
- Iannaccone S.T., Bove K.E., Vogler C., Azzarelli B., Muller J. Muscle maturation delay in infantile myotonic dystrophy. Arch. Pathol. Lab. Med. 1986;110:405–411. - PubMed
-
- Pelé M., Tiret L., Kessler J.L., Blot S., Panthier J.J. SINE Exonic insertion in the PTPLA gene leads to multiple splicing defects and segregates with the autosomal recessive centronuclear myopathy in dogs. Hum. Mol. Genet. 2005;14:1417–1427. doi:10.1093/hmg/ddi151. - DOI - PubMed
-
- Maquat L.E. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol. 2004;5:89–99. doi:10.1038/nrm1310. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
