

Genome-wide prediction of traits with different genetic architecture through efficient variable selection
- PMID: 23934883
- PMCID: PMC3781982
- DOI: 10.1534/genetics.113.150078
Genome-wide prediction of traits with different genetic architecture through efficient variable selection
Abstract
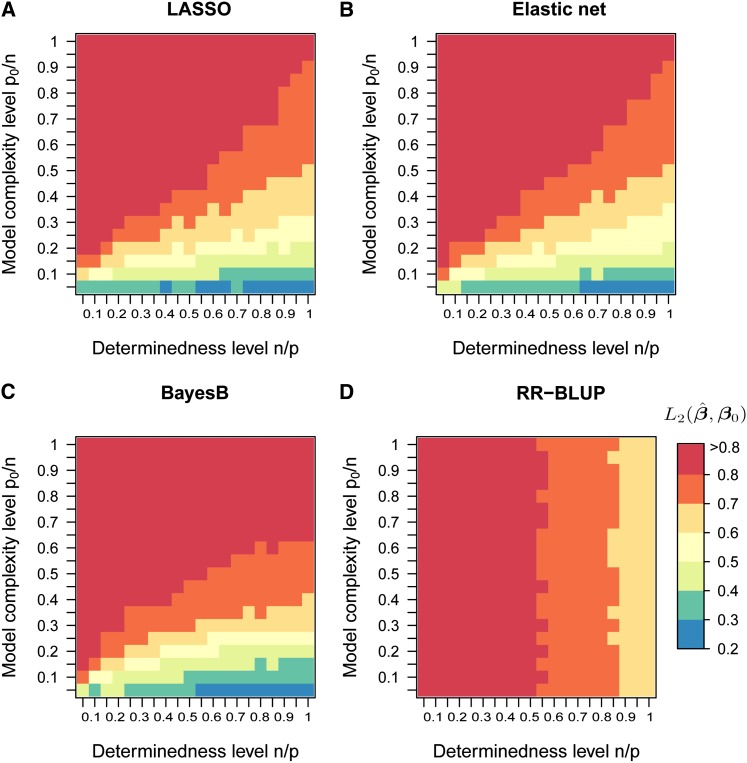
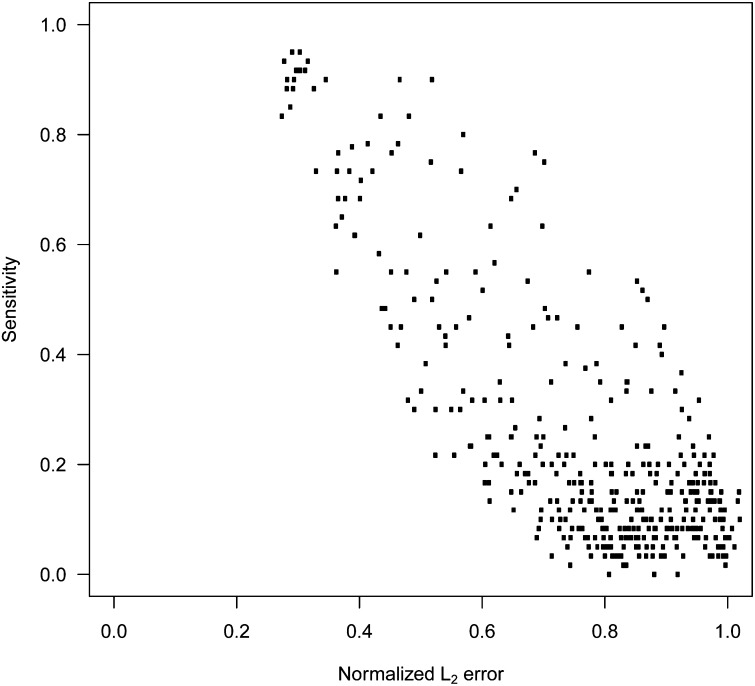
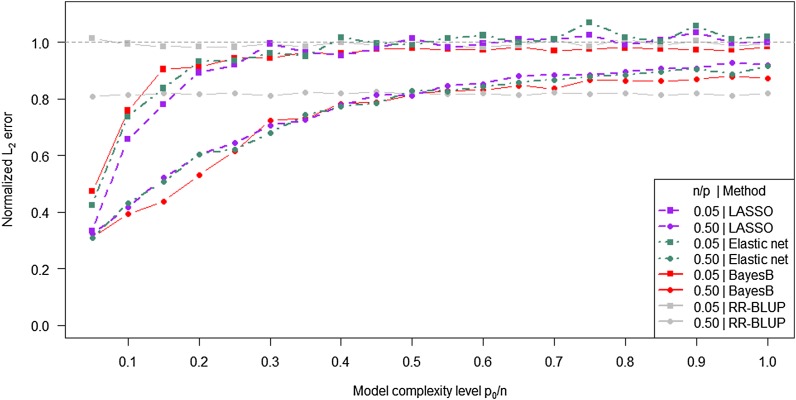
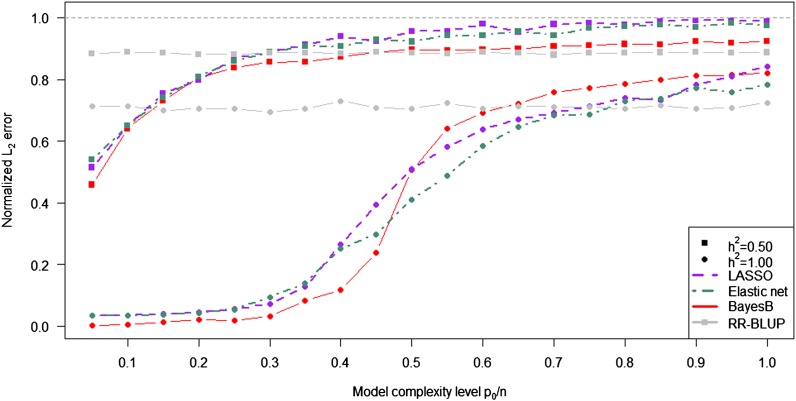
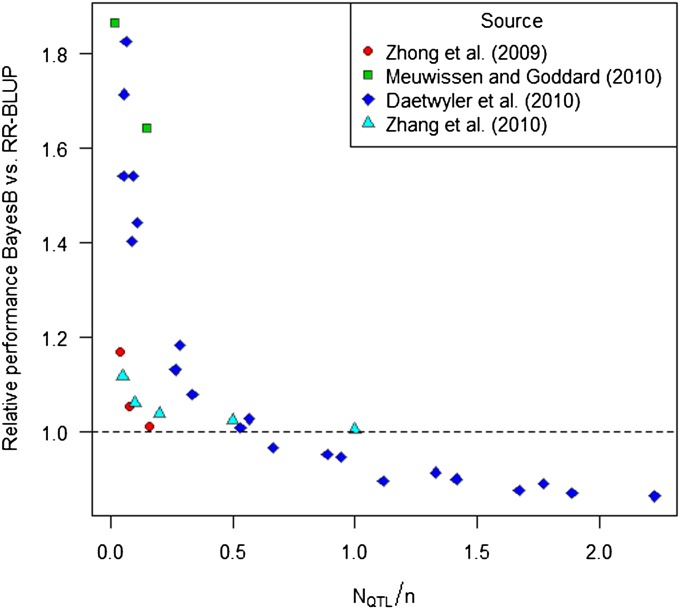
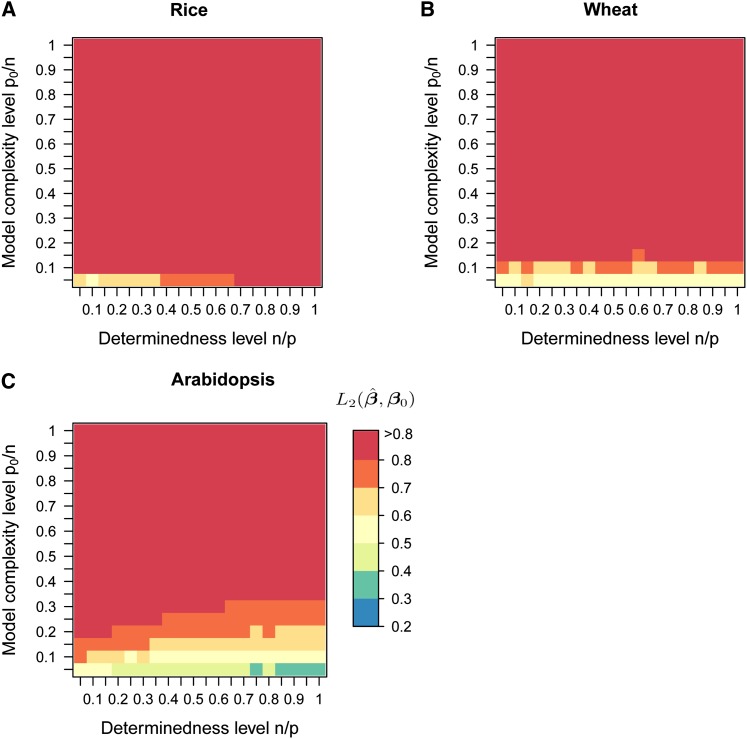
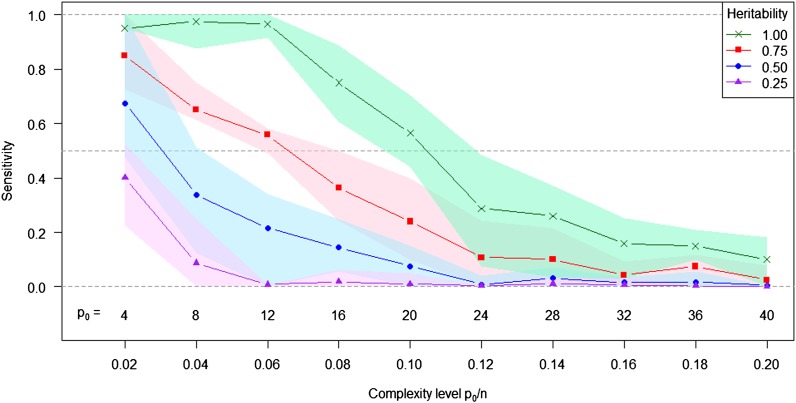
In genome-based prediction there is considerable uncertainty about the statistical model and method required to maximize prediction accuracy. For traits influenced by a small number of quantitative trait loci (QTL), predictions are expected to benefit from methods performing variable selection [e.g., BayesB or the least absolute shrinkage and selection operator (LASSO)] compared to methods distributing effects across the genome [ridge regression best linear unbiased prediction (RR-BLUP)]. We investigate the assumptions underlying successful variable selection by combining computer simulations with large-scale experimental data sets from rice (Oryza sativa L.), wheat (Triticum aestivum L.), and Arabidopsis thaliana (L.). We demonstrate that variable selection can be successful when the number of phenotyped individuals is much larger than the number of causal mutations contributing to the trait. We show that the sample size required for efficient variable selection increases dramatically with decreasing trait heritabilities and increasing extent of linkage disequilibrium (LD). We contrast and discuss contradictory results from simulation and experimental studies with respect to superiority of variable selection methods over RR-BLUP. Our results demonstrate that due to long-range LD, medium heritabilities, and small sample sizes, superiority of variable selection methods cannot be expected in plant breeding populations even for traits like FRIGIDA gene expression in Arabidopsis and flowering time in rice, assumed to be influenced by a few major QTL. We extend our conclusions to the analysis of whole-genome sequence data and infer upper bounds for the number of causal mutations which can be identified by LASSO. Our results have major impact on the choice of statistical method needed to make credible inferences about genetic architecture and prediction accuracy of complex traits.
Keywords: GenPred; complex traits; genetic architecture; genome-based prediction; plant breeding populations; shared data resources; variable selection.
Figures
References
-
- Albrecht T., Wimmer V., Auinger H.-J., Erbe M., Knaak C., et al. , 2011. Genome-based prediction of testcross values in maize. Theor. Appl. Genet. 123: 339–350. - PubMed
-
- Bates, D., and M. Maechler, 2012 Matrix: sparse and dense matrix classes and methods. R Package Version 1.0–6.
-
- Butts C. T., 2008. Network: a package for managing relational data in R. J. Stat. Softw. 24: 1–36.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
