

A novel approach to estimating heterozygosity from low-coverage genome sequence
- PMID: 23934885
- PMCID: PMC3781980
- DOI: 10.1534/genetics.113.154500
A novel approach to estimating heterozygosity from low-coverage genome sequence
Abstract
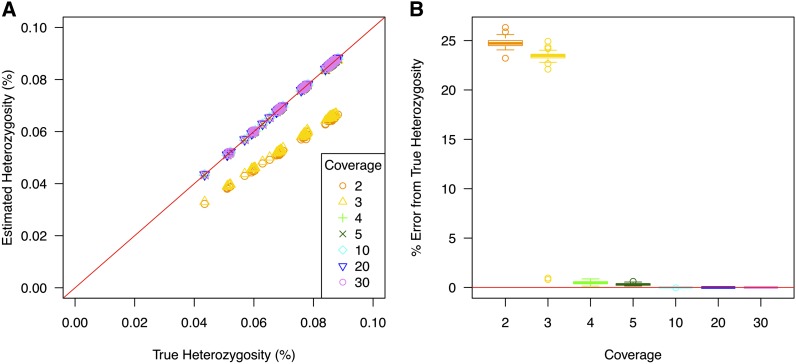
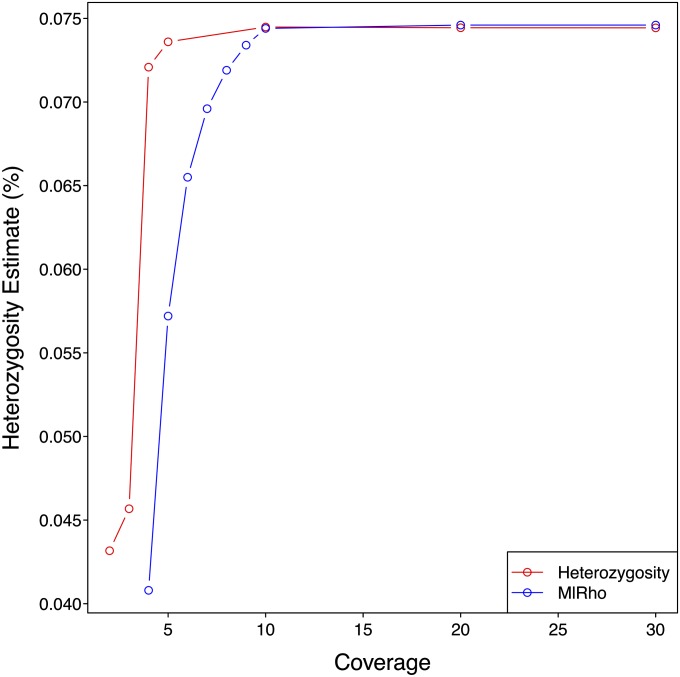
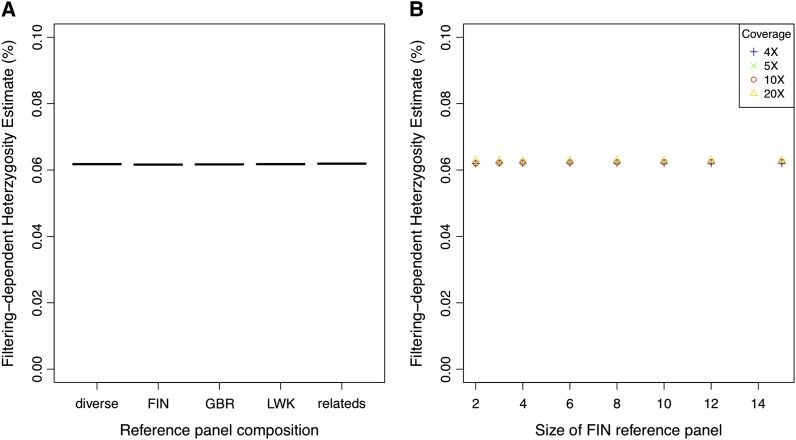
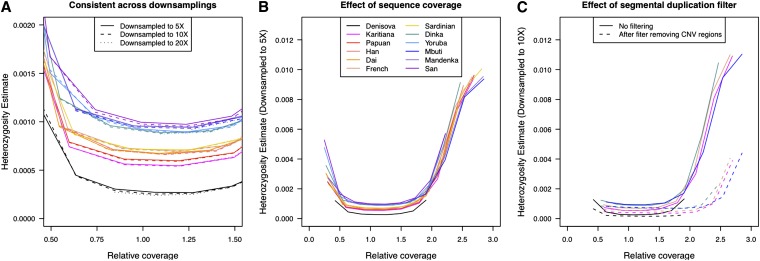
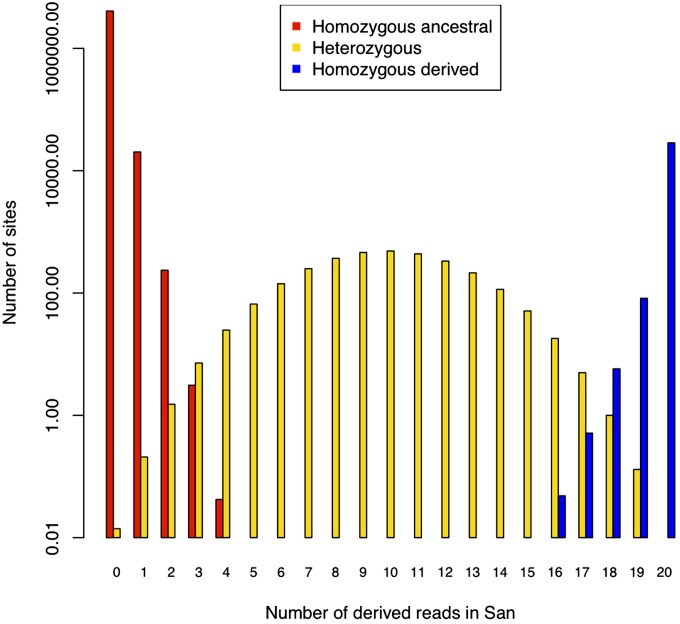
High-throughput shotgun sequence data make it possible in principle to accurately estimate population genetic parameters without confounding by SNP ascertainment bias. One such statistic of interest is the proportion of heterozygous sites within an individual's genome, which is informative about inbreeding and effective population size. However, in many cases, the available sequence data of an individual are limited to low coverage, preventing the confident calling of genotypes necessary to directly count the proportion of heterozygous sites. Here, we present a method for estimating an individual's genome-wide rate of heterozygosity from low-coverage sequence data, without an intermediate step that calls genotypes. Our method jointly learns the shared allele distribution between the individual and a panel of other individuals, together with the sequencing error distributions and the reference bias. We show our method works well, first, by its performance on simulated sequence data and, second, on real sequence data where we obtain estimates using low-coverage data consistent with those from higher coverage. We apply our method to obtain estimates of the rate of heterozygosity for 11 humans from diverse worldwide populations and through this analysis reveal the complex dependency of local sequencing coverage on the true underlying heterozygosity, which complicates the estimation of heterozygosity from sequence data. We show how we can use filters to correct for the confounding arising from sequencing depth. We find in practice that ratios of heterozygosity are more interpretable than absolute estimates and show that we obtain excellent conformity of ratios of heterozygosity with previous estimates from higher-coverage data.
Keywords: heterozygosity; low-coverage sequence data.
Figures
References
-
- Jakobsson M., Scholz S. W., Scheet P., Gibbs J. R., VanLiere J. M., et al. , 2008. Genotype, haplotype and copy-number variation in worldwide human populations. Nature 451: 998–1003. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
