

Oxidative stress and replication-independent DNA breakage induced by arsenic in Saccharomyces cerevisiae
- PMID: 23935510
- PMCID: PMC3723488
- DOI: 10.1371/journal.pgen.1003640
Oxidative stress and replication-independent DNA breakage induced by arsenic in Saccharomyces cerevisiae
Abstract
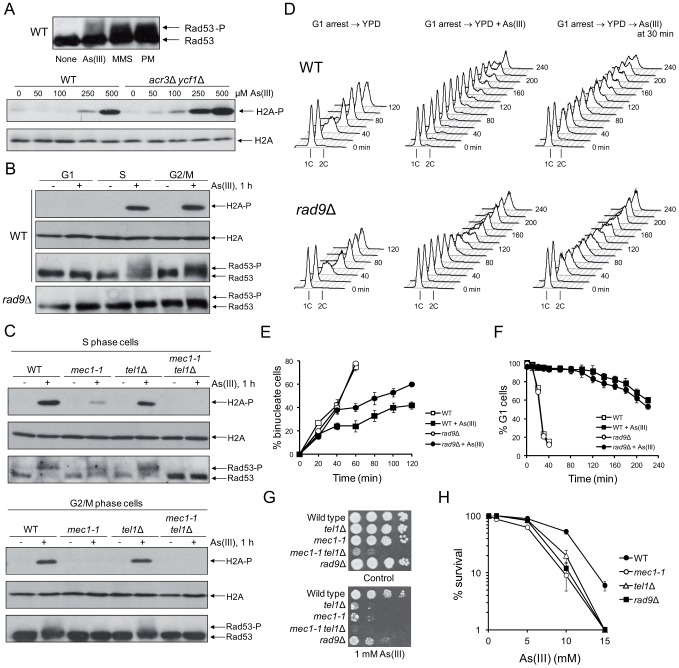
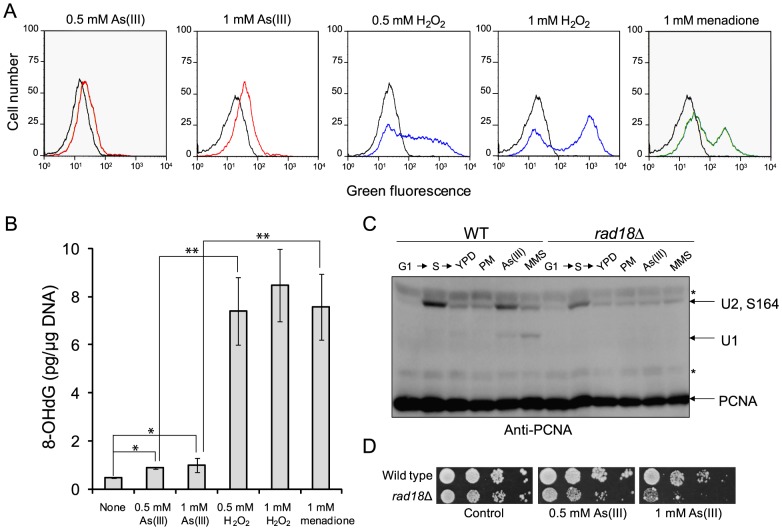
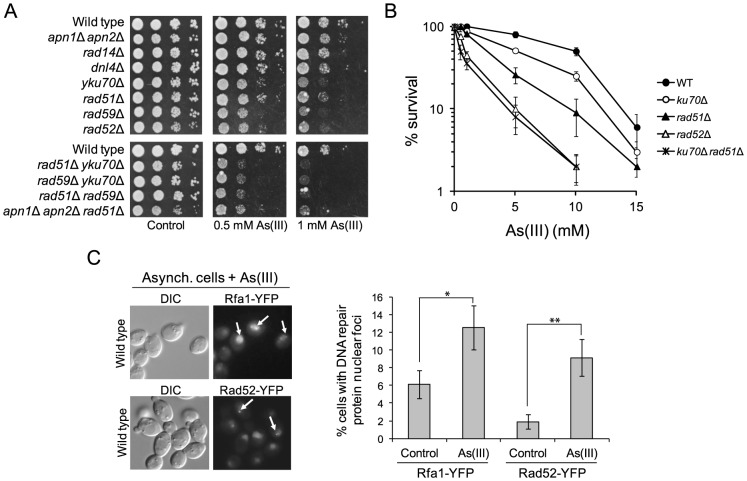
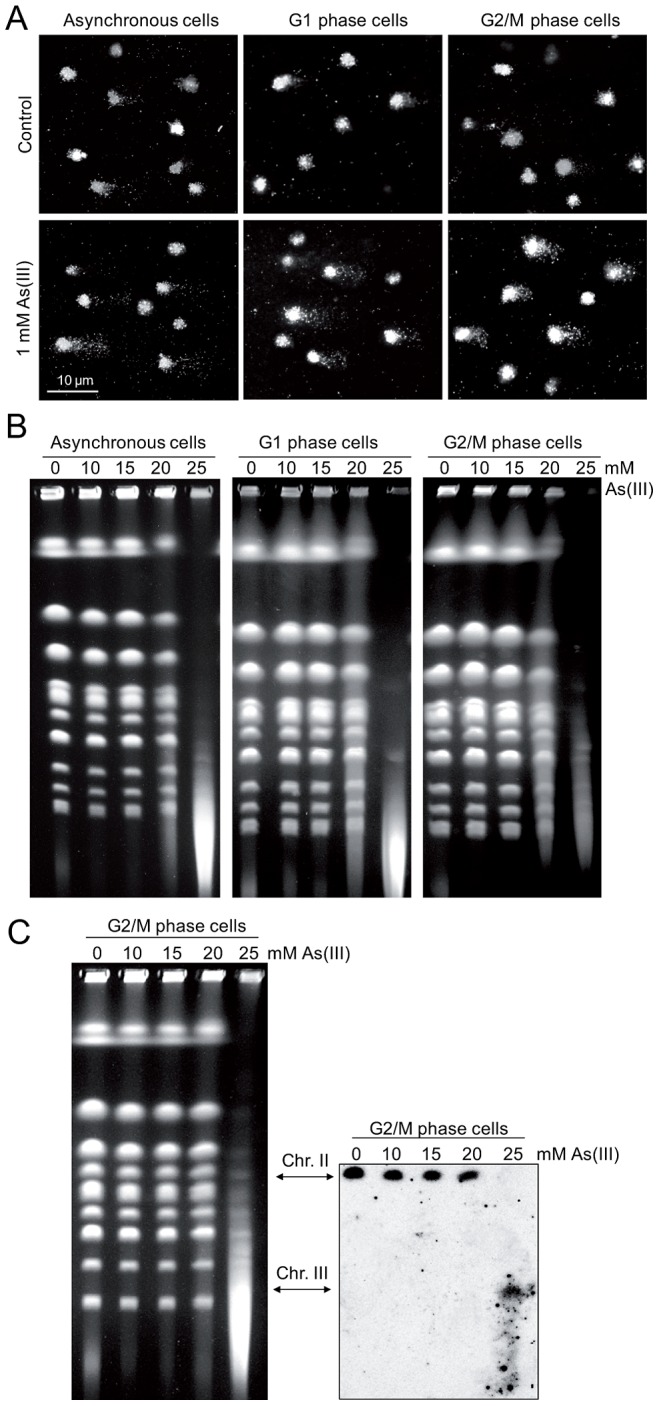
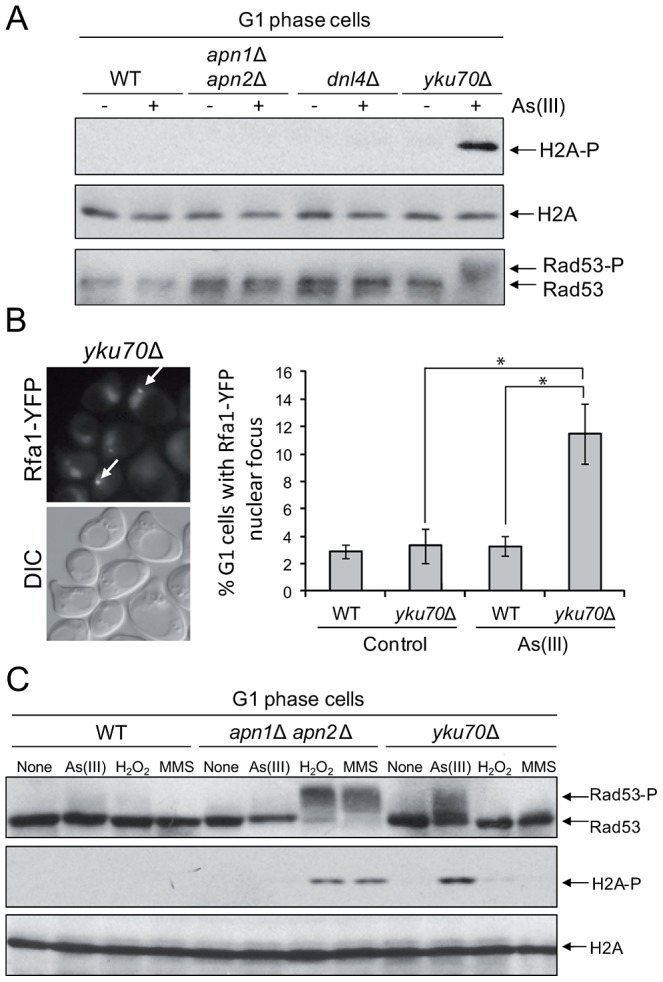
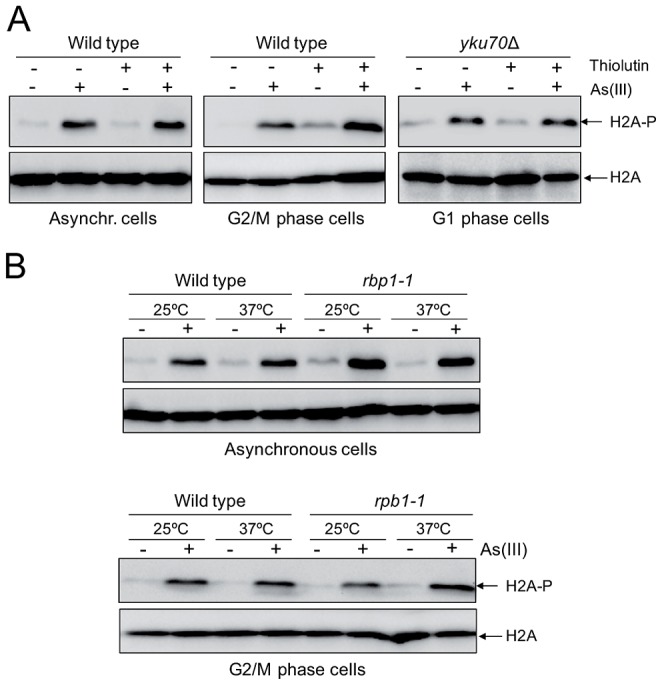
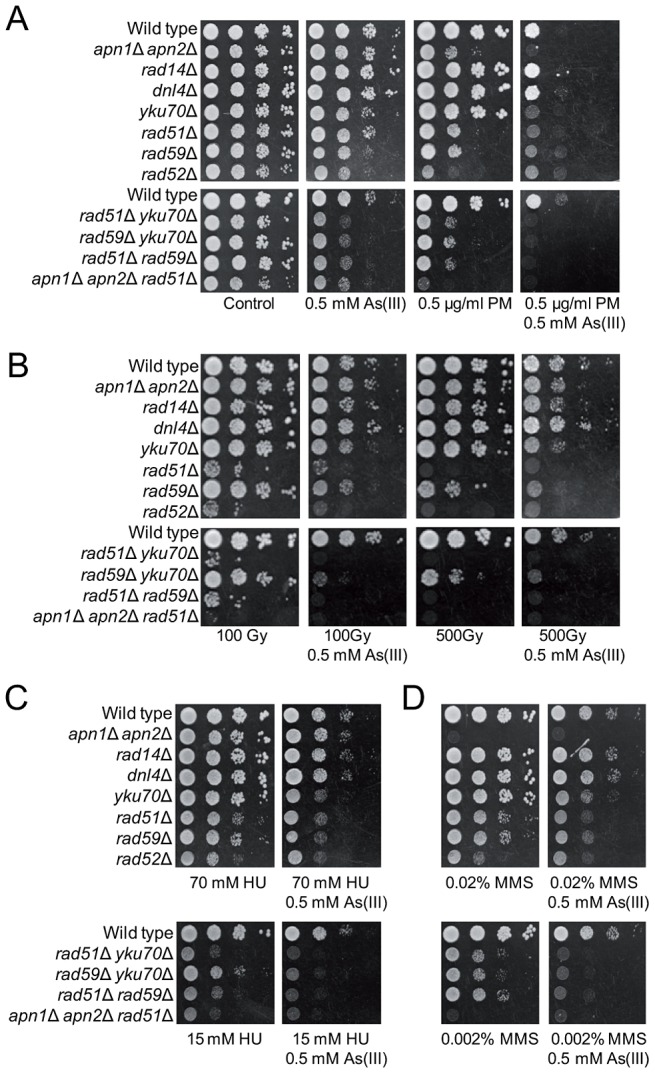
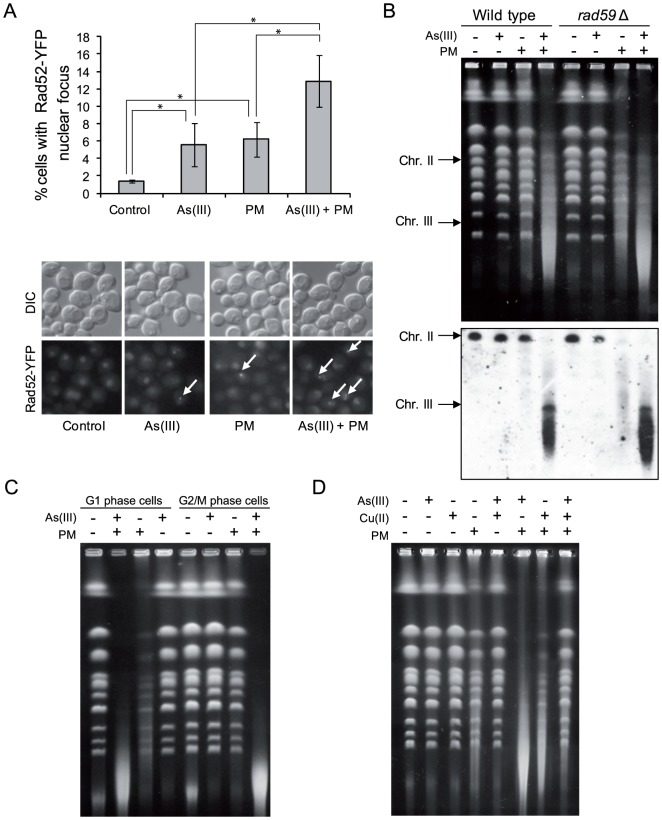
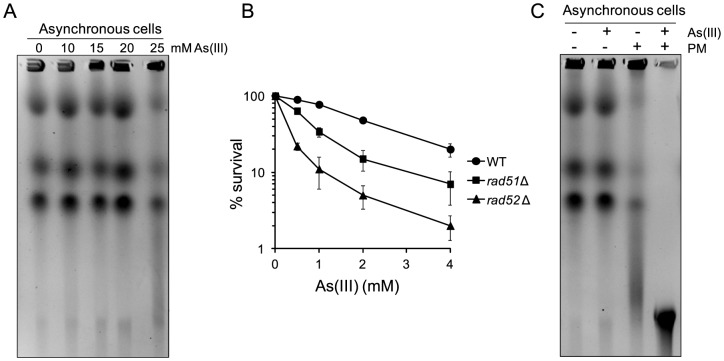
Arsenic is a well-established human carcinogen of poorly understood mechanism of genotoxicity. It is generally accepted that arsenic acts indirectly by generating oxidative DNA damage that can be converted to replication-dependent DNA double-strand breaks (DSBs), as well as by interfering with DNA repair pathways and DNA methylation. Here we show that in budding yeast arsenic also causes replication and transcription-independent DSBs in all phases of the cell cycle, suggesting a direct genotoxic mode of arsenic action. This is accompanied by DNA damage checkpoint activation resulting in cell cycle delays in S and G2/M phases in wild type cells. In G1 phase, arsenic activates DNA damage response only in the absence of the Yku70-Yku80 complex which normally binds to DNA ends and inhibits resection of DSBs. This strongly indicates that DSBs are produced by arsenic in G1 but DNA ends are protected by Yku70-Yku80 and thus invisible for the checkpoint response. Arsenic-induced DSBs are processed by homologous recombination (HR), as shown by Rfa1 and Rad52 nuclear foci formation and requirement of HR proteins for cell survival during arsenic exposure. We show further that arsenic greatly sensitizes yeast to phleomycin as simultaneous treatment results in profound accumulation of DSBs. Importantly, we observed a similar response in fission yeast Schizosaccharomyces pombe, suggesting that the mechanisms of As(III) genotoxicity may be conserved in other organisms.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Tapio S, Grosche B (2006) Arsenic in the aetiology of cancer. Mutat Res 612: 215–246. - PubMed
-
- Dilda PJ, Hogg PJ (2007) Arsenical-based cancer drugs. Cancer Treat Rev 33: 542–564. - PubMed
-
- Murray HW, Berman JD, Davies CR, Saravia NG (2005) Advances in leishmaniasis. Lancet 366: 1561–1577. - PubMed
-
- Liu JX, Zhou GB, Chen SJ, Chen Z (2012) Arsenic compounds: revived ancient remedies in the fight against human malignancies. Curr Opin Chem Biol 16: 92–98. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
