

Functional characterisation of alpha-galactosidase a mutations as a basis for a new classification system in fabry disease
- PMID: 23935525
- PMCID: PMC3731228
- DOI: 10.1371/journal.pgen.1003632
Functional characterisation of alpha-galactosidase a mutations as a basis for a new classification system in fabry disease
Abstract
Fabry disease (FD) is an X-linked hereditary defect of glycosphingolipid storage caused by mutations in the gene encoding the lysosomal hydrolase α-galactosidase A (GLA, α-gal A). To date, over 400 mutations causing amino acid substitutions have been described. Most of these mutations are related to the classical Fabry phenotype. Generally in lysosomal storage disorders a reliable genotype/phenotype correlation is difficult to achieve, especially in FD with its X-linked mode of inheritance. In order to predict the metabolic consequence of a given mutation, we combined in vitro enzyme activity with in vivo biomarker data. Furthermore, we used the pharmacological chaperone (PC) 1-deoxygalactonojirimycin (DGJ) as a tool to analyse the influence of individual mutations on subcellular organelle-trafficking and stability. We analysed a significant number of mutations and correlated the obtained properties to the clinical manifestation related to the mutation in order to improve our knowledge of the identity of functional relevant amino acids. Additionally, we illustrate the consequences of different mutations on plasma lyso-globotriaosylsphingosine (lyso-Gb3) accumulation in the patients' plasma, a biomarker proven to reflect the impaired substrate clearance caused by specific mutations. The established system enables us to provide information for the clinical relevance of PC therapy for a given mutant. Finally, in order to generate reliable predictions of mutant GLA defects we compared the different data sets to reveal the most coherent system to reflect the clinical situation.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
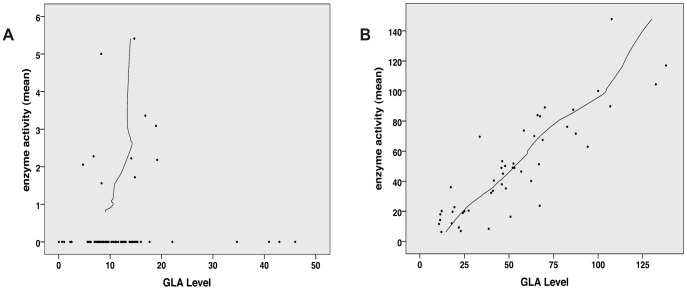
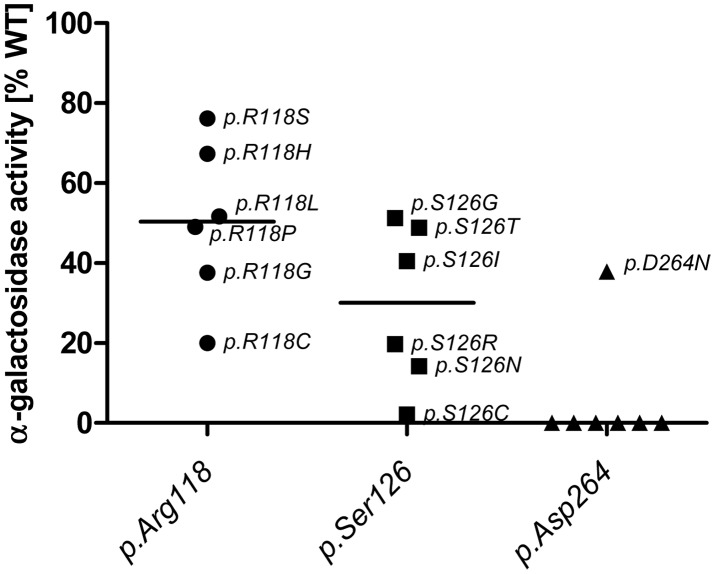
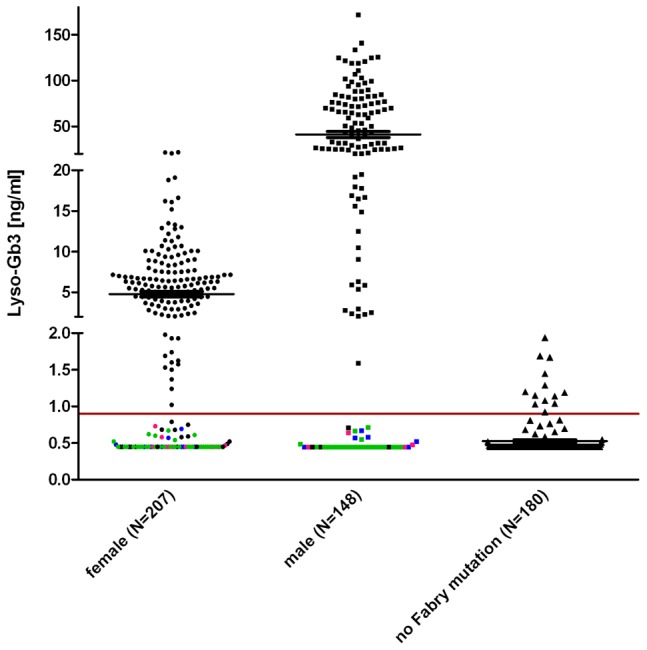
References
-
- Ishii S, Chang HH, Kawasaki K, Yasuda K, Wu HL, et al. (2007) Mutant α-galactosidase A enzymes identified in Fabry disease patients with residual enzyme activity: biochemical characterization and restoration of normal intracellular processing by 1-deoxygalactonojirimycin. Biochem J 406 Pt 2: 285–295. - PMC - PubMed
-
- Kotanko P, Kramar R, Devrnja D, Paschke E, Voigtländer T, et al. (2004) Results of a Nationwide Screening for Anderson-Fabry Disease among Dialysis Patients. J Am Soc Nephrol 15: 1323–1329. - PubMed
-
- Mehta A, Beck M, Eyskens F, Feliciani C, Kantola I, et al. (2010) Fabry disease: a review of current management strategies. QJM 103: 641–59. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
