

Orthosteric binding of ρ-Da1a, a natural peptide of snake venom interacting selectively with the α1A-adrenoceptor
- PMID: 23935897
- PMCID: PMC3723878
- DOI: 10.1371/journal.pone.0068841
Orthosteric binding of ρ-Da1a, a natural peptide of snake venom interacting selectively with the α1A-adrenoceptor
Abstract
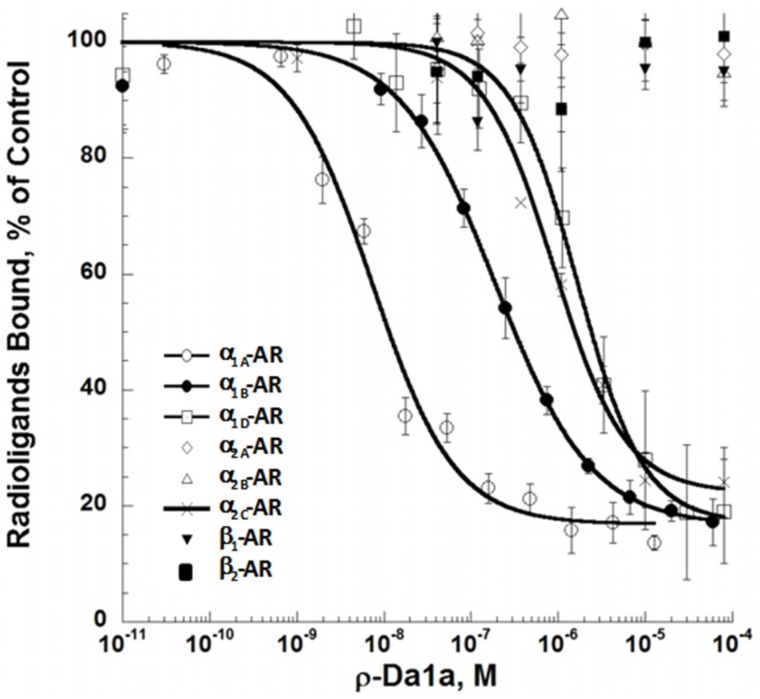
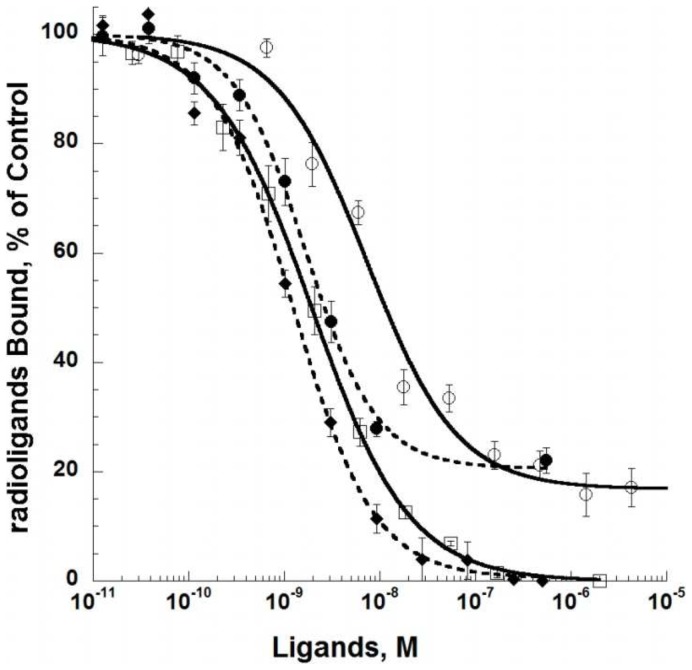
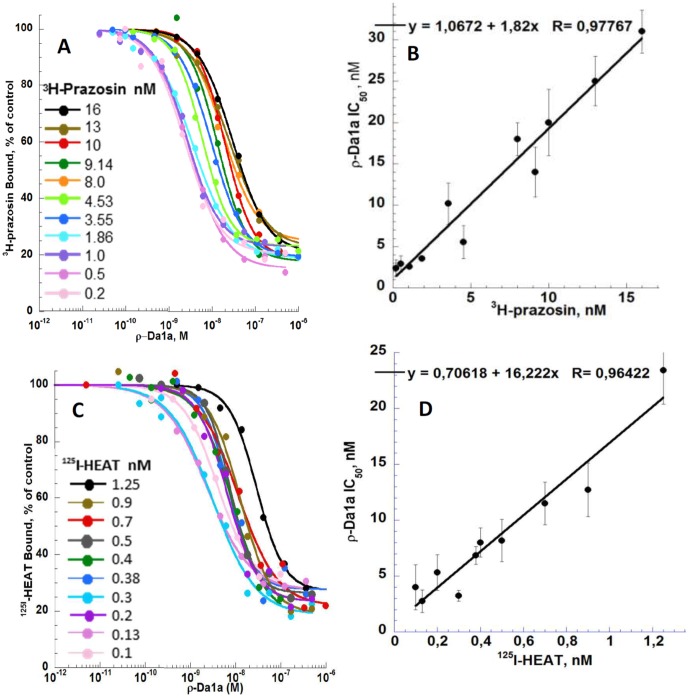
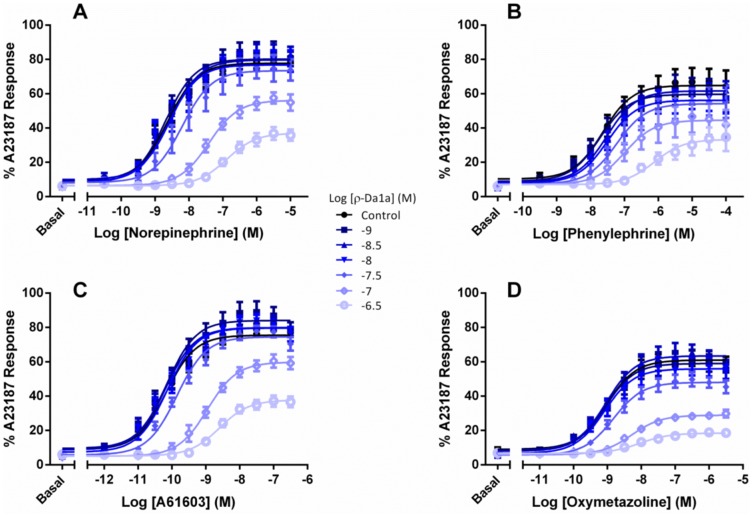
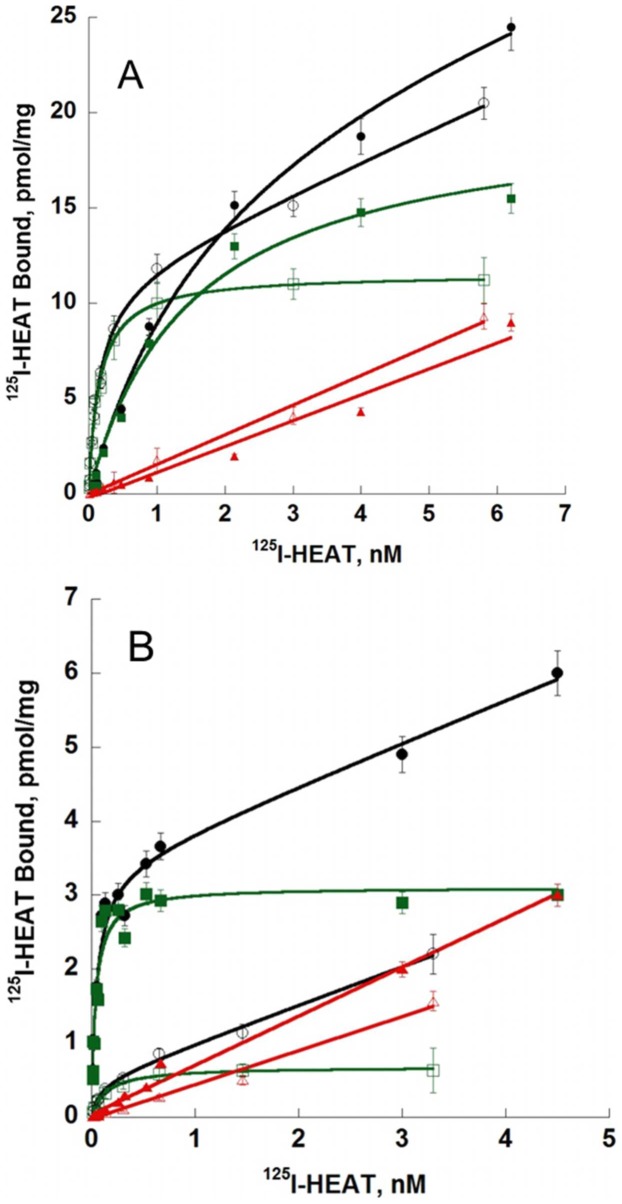
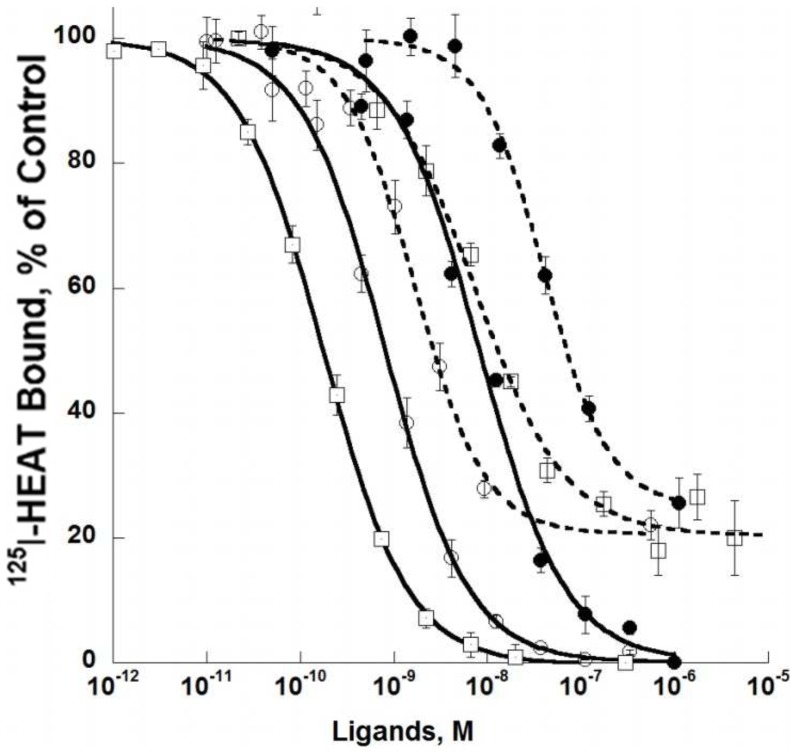
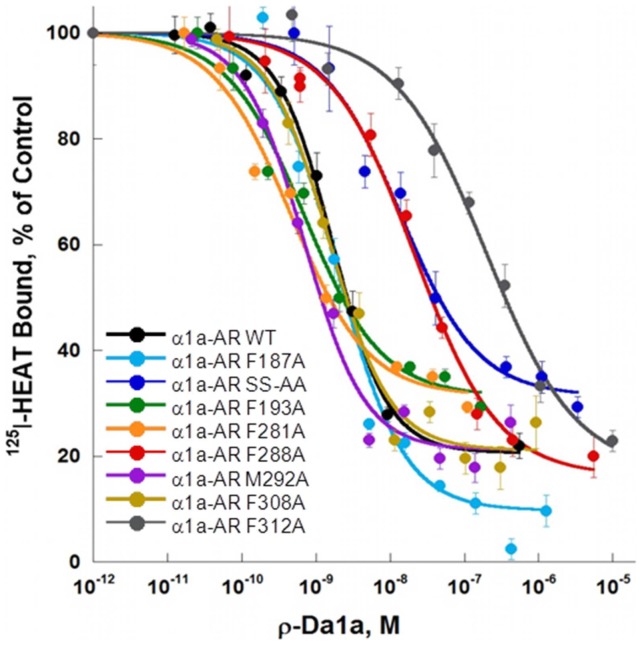
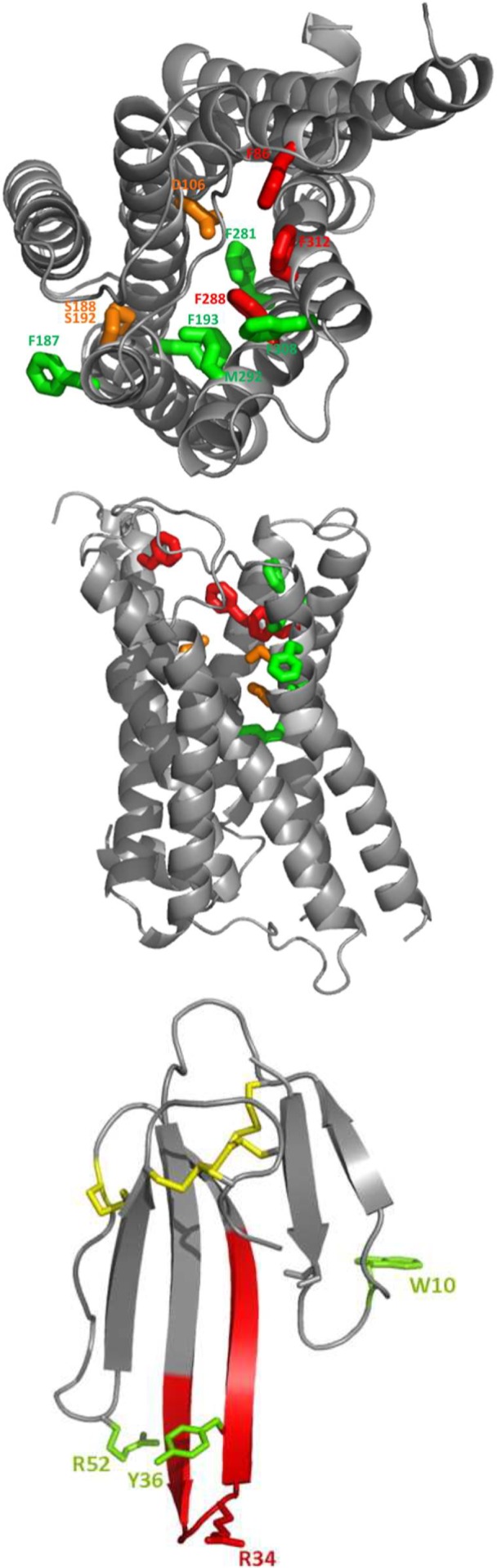
ρ-Da1a is a three-finger fold toxin from green mamba venom that is highly selective for the α1A-adrenoceptor. This toxin has atypical pharmacological properties, including incomplete inhibition of (3)H-prazosin or (125)I-HEAT binding and insurmountable antagonist action. We aimed to clarify its mode of action at the α1A-adrenoceptor. The affinity (pKi 9.26) and selectivity of ρ-Da1a for the α1A-adrenoceptor were confirmed by comparing binding to human adrenoceptors expressed in eukaryotic cells. Equilibrium and kinetic binding experiments were used to demonstrate that ρ-Da1a, prazosin and HEAT compete at the α1A-adrenoceptor. ρ-Da1a did not affect the dissociation kinetics of (3)H-prazosin or (125)I-HEAT, and the IC50 of ρ-Da1a, determined by competition experiments, increased linearly with the concentration of radioligands used, while the residual binding by ρ-Da1a remained stable. The effect of ρ-Da1a on agonist-stimulated Ca(2+) release was insurmountable in the presence of phenethylamine- or imidazoline-type agonists. Ten mutations in the orthosteric binding pocket of the α1A-adrenoceptor were evaluated for alterations in ρ-Da1a affinity. The D106(3.32)A and the S188(5.42)A/S192(5.46)A receptor mutations reduced toxin affinity moderately (6 and 7.6 times, respectively), while the F86(2.64)A, F288(6.51)A and F312(7.39)A mutations diminished it dramatically by 18- to 93-fold. In addition, residue F86(2.64) was identified as a key interaction point for (125)I-HEAT, as the variant F86(2.64)A induced a 23-fold reduction in HEAT affinity. Unlike the M1 muscarinic acetylcholine receptor toxin MT7, ρ-Da1a interacts with the human α1A-adrenoceptor orthosteric pocket and shares receptor interaction points with antagonist (F86(2.64), F288(6.51) and F312(7.39)) and agonist (F288(6.51) and F312(7.39)) ligands. Its selectivity for the α1A-adrenoceptor may result, at least partly, from its interaction with the residue F86(2.64), which appears to be important also for HEAT binding.
Conflict of interest statement
Figures

References
-
- Escoubas P, King GF (2009) Venomics as a drug discovery platform. Expert Rev Proteomics 6: 221–224. - PubMed
-
- Halai R, Craik DJ (2009) Conotoxins: natural product drug leads. Nat Prod Rep 26: 526–536. - PubMed
-
- Lewis RJ, Garcia ML (2003) Therapeutic potential of venom peptides. Nat Rev Drug Discov 2: 790–802. - PubMed
-
- Shen GS, Layer RT, McCabe RT (2000) Conopeptides: From deadly venoms to novel therapeutics. Drug Discov Today 5: 98–106. - PubMed
-
- Maïga A, Mourier G, Quinton L, Rouget C, Gales C, et al. (2012) G protein-coupled receptors, an unexploited animal toxin targets: Exploration of green mamba venom for novel drug candidates active against adrenoceptors. Toxicon 59: 487–496. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
