

Neurodegeneration in Friedreich's ataxia: from defective frataxin to oxidative stress
- PMID: 23936609
- PMCID: PMC3725840
- DOI: 10.1155/2013/487534
Neurodegeneration in Friedreich's ataxia: from defective frataxin to oxidative stress
Abstract
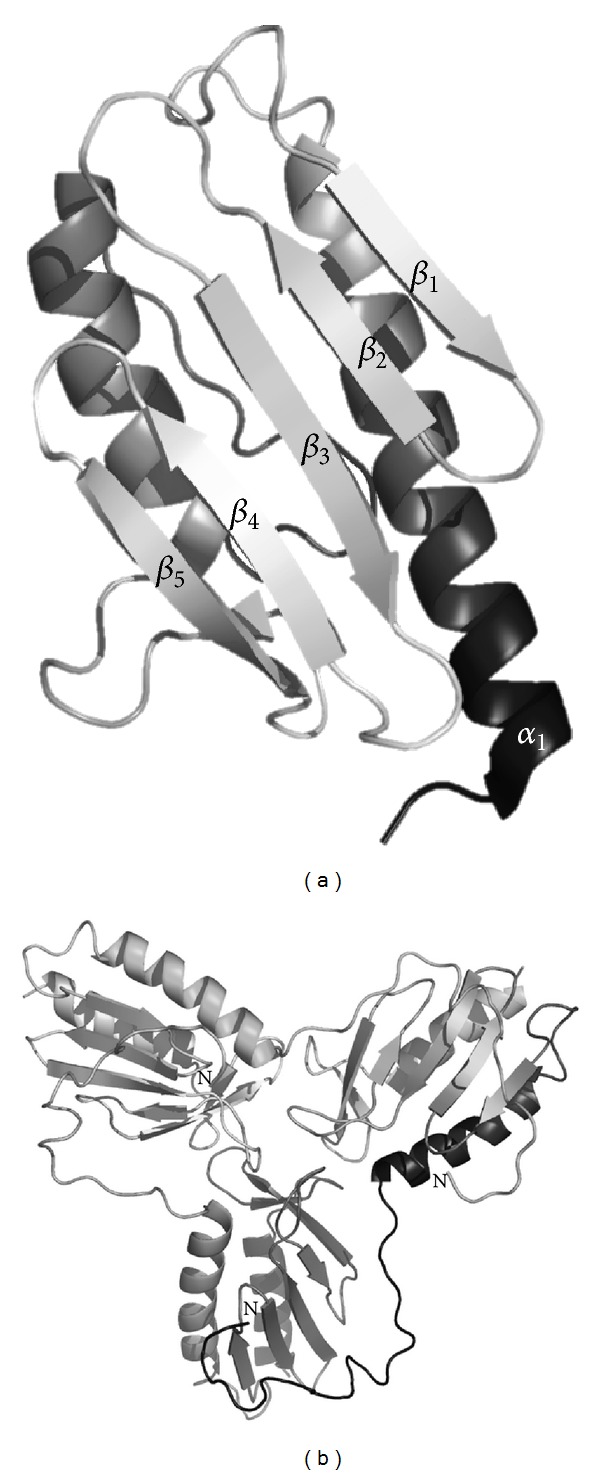
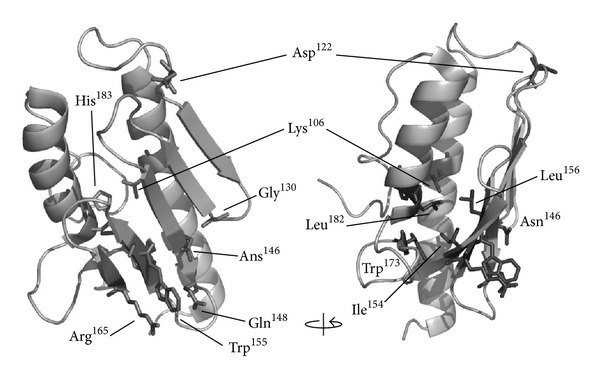
Friedreich's ataxia is the most common inherited autosomal recessive ataxia and is characterized by progressive degeneration of the peripheral and central nervous systems and cardiomyopathy. This disease is caused by the silencing of the FXN gene and reduced levels of the encoded protein, frataxin. Frataxin is a mitochondrial protein that functions primarily in iron-sulfur cluster synthesis. This small protein with an α / β sandwich fold undergoes complex processing and imports into the mitochondria, generating isoforms with distinct N-terminal lengths which may underlie different functionalities, also in respect to oligomerization. Missense mutations in the FXN coding region, which compromise protein folding, stability, and function, are found in 4% of FRDA heterozygous patients and are useful to understand how loss of functional frataxin impacts on FRDA physiopathology. In cells, frataxin deficiency leads to pleiotropic phenotypes, including deregulation of iron homeostasis and increased oxidative stress. Increasing amount of data suggest that oxidative stress contributes to neurodegeneration in Friedreich's ataxia.
Figures
References
-
- Friedreich N. Über degenerative Atrophie der spinalen Hinterstränge. Virchows Archiv fur Pathologische Anatomie und Physiologie und für klinische Medizin. 1863;27:1–26.
-
- Friedreich N. Über degenerative Atrophie der spinalen Hinterstränge. Virchows Archiv fur Pathologische Anatomie und Physiologie und für klinische Medizin. 1863;26:391–419.
-
- Friedreich N. Über degenerative Atrophie der spinalen Hinterstränge. Virchows Archiv fur Pathologische Anatomie und Physiologie und für klinische Medizin. 1863;26:433–459.
-
- Harding AE. Friedreich’s ataxia: a clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain. 1981;104(3):589–620. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
