

On the versatility of von Willebrand factor
- PMID: 23936617
- PMCID: PMC3736882
- DOI: 10.4084/MJHID.2013.046
On the versatility of von Willebrand factor
Abstract
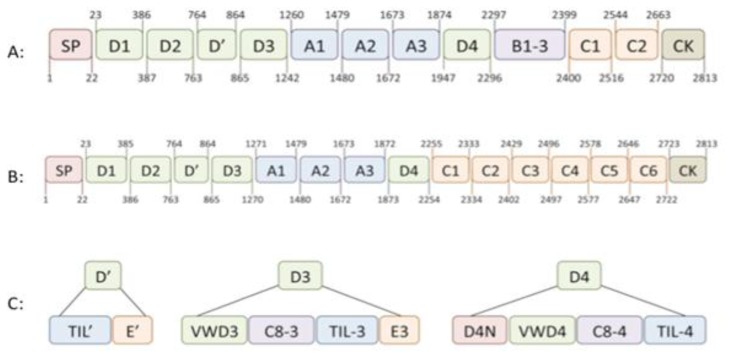
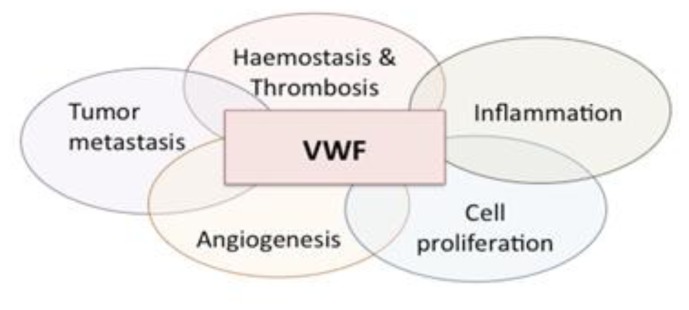
Von Willebrand factor (VWF) is a large multimeric protein, the function of which has been demonstrated to be pivotal to the haemostatic system. Indeed, quantitative and/or qualitative abnormalities of VWF are associated with the bleeding disorder Von Willebrand disease (VWD). Moreover, increased plasma concentrations of VWF have been linked to an increased risk for thrombotic complications. In the previous decades, many studies have contributed to our understanding of how VWF is connected to the haemostatic system, particularly with regard to structure-function relationships. Interactive sites for important ligands of VWF (such as factor VIII, collagen, glycoprotein Ibα, integrin αIIbβ3 and protease ADAMTS13) have been identified, and mutagenesis studies have confirmed the physiological relevance of the interactions between VWF and these ligands. However, we have also become aware that VWF has a more versatile character than previously thought, given its potential role in various non-hemostatic processes, like intimal thickening, tumor cell apoptosis and inflammatory processes. In the presence review, a summary of our knowledge on VWF structure-function relationships is provided in the context of the "classical" haemostatic task of VWF and in perspective of pathological processes beyond haemostasis.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous