

Transcriptome deep-sequencing and clustering of expressed isoforms from Favia corals
- PMID: 23937070
- PMCID: PMC3751062
- DOI: 10.1186/1471-2164-14-546
Transcriptome deep-sequencing and clustering of expressed isoforms from Favia corals
Abstract
Background: Genomic and transcriptomic sequence data are essential tools for tackling ecological problems. Using an approach that combines next-generation sequencing, de novo transcriptome assembly, gene annotation and synthetic gene construction, we identify and cluster the protein families from Favia corals from the northern Red Sea.
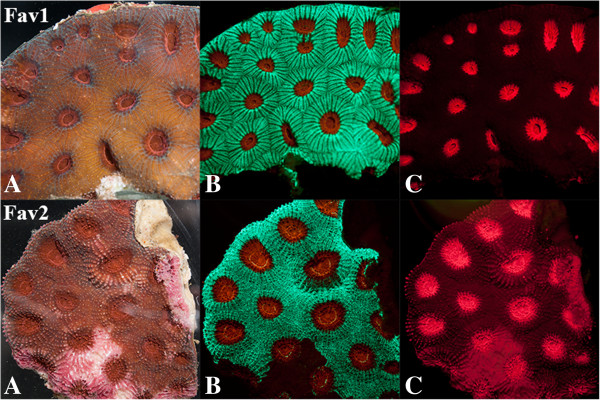
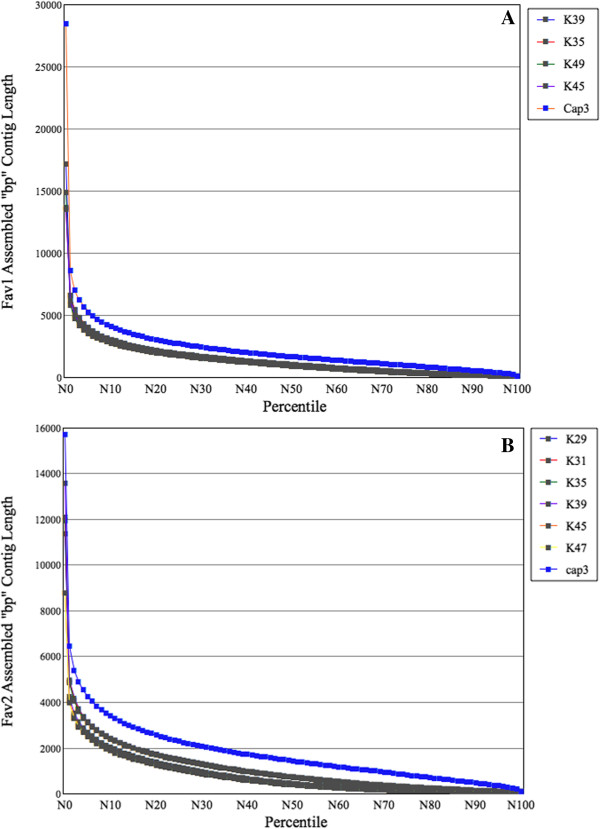
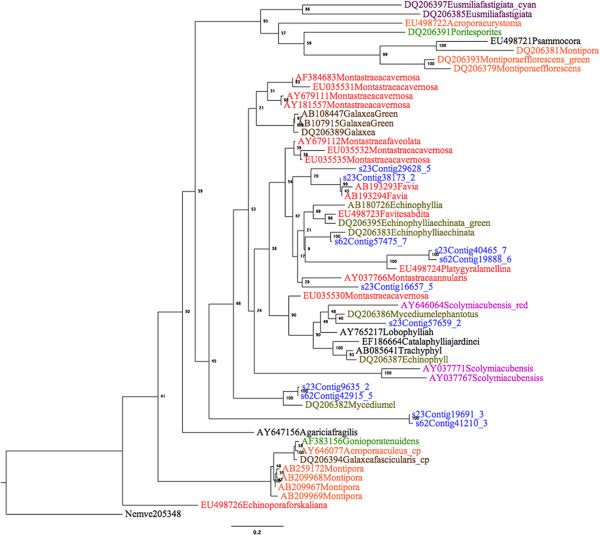
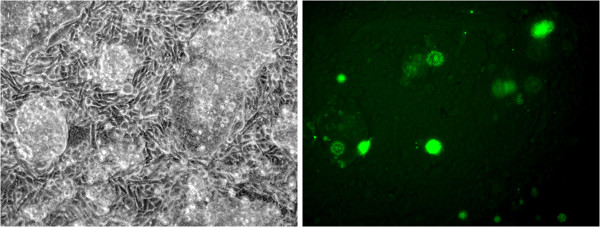
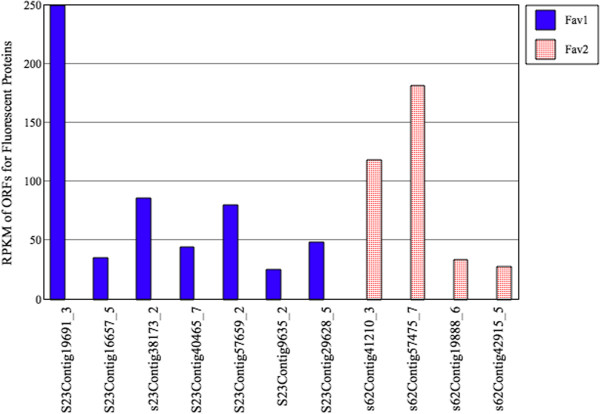
Results: We obtained 80 million 75 bp paired-end cDNA reads from two Favia adult samples collected at 65 m (Fav1, Fav2) on the Illumina GA platform, and generated two de novo assemblies using ABySS and CAP3. After removing redundancy and filtering out low quality reads, our transcriptome datasets contained 58,268 (Fav1) and 62,469 (Fav2) contigs longer than 100 bp, with N50 values of 1,665 bp and 1,439 bp, respectively. Using the proteome of the sea anemone Nematostella vectensis as a reference, we were able to annotate almost 20% of each dataset using reciprocal homology searches. Homologous clustering of these annotated transcripts allowed us to divide them into 7,186 (Fav1) and 6,862 (Fav2) homologous transcript clusters (E-value ≤ 2e(-30)). Functional annotation categories were assigned to homologous clusters using the functional annotation of Nematostella vectensis. General annotation of the assembled transcripts was improved 1-3% using the Acropora digitifera proteome. In addition, we screened these transcript isoform clusters for fluorescent proteins (FPs) homologs and identified seven potential FP homologs in Fav1, and four in Fav2. These transcripts were validated as bona fide FP transcripts via robust fluorescence heterologous expression. Annotation of the assembled contigs revealed that 1.34% and 1.61% (in Fav1 and Fav2, respectively) of the total assembled contigs likely originated from the corals' algal symbiont, Symbiodinium spp.
Conclusions: Here we present a study to identify the homologous transcript isoform clusters from the transcriptome of Favia corals using a far-related reference proteome. Furthermore, the symbiont-derived transcripts were isolated from the datasets and their contribution quantified. This is the first annotated transcriptome of the genus Favia, a major increase in genomics resources available in this important family of corals.
Figures

References
-
- Metzker ML. Sequencing technologies—the next generation. Nat Rev Gen. 2009;11(1):31–46. - PubMed
-
- Elmer KR, Fan S, Gunter HM, Jones JC, Boekhoff S, Kuraku S, Meyer A. Rapid evolution and selection inferred from the transcriptomes of sympatric crater lake cichlid fishes. Mole Ecol. 2010;19(Suppl 1):197–211. - PubMed
-
- Yang SS, Tu ZJ, Cheung F, Xu WW, Lamb JF, Jung HJ, Vance CP, Gronwald JW. Using RNA-Seq for gene identification, polymorphism detection and transcript profiling in two alfalfa genotypes with divergent cell wall composition in stems. BMC Genomics. 2011;12:199. doi: 10.1186/1471-2164-12-199. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
