

Spinal muscular atrophy: development and implementation of potential treatments
- PMID: 23939659
- PMCID: PMC3876415
- DOI: 10.1002/ana.23995
Spinal muscular atrophy: development and implementation of potential treatments
Abstract
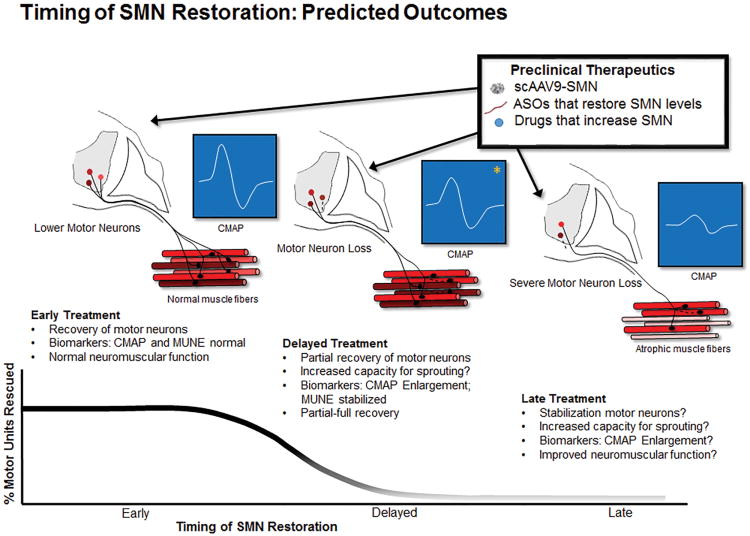
In neurodegenerative disorders, effective treatments are urgently needed, along with methods to determine whether treatment worked. In this review, we discuss the rapid progress in the understanding of recessive proximal spinal muscular atrophy and how this is leading to exciting potential treatments of the disease. Spinal muscular atrophy is caused by loss of the survival motor neuron 1 (SMN1) gene and reduced levels of SMN protein. The critical downstream targets of SMN deficiency that result in motor neuron loss are not known. However, increasing SMN levels has a marked impact in mouse models, and these therapeutics are rapidly moving toward clinical trials. Promising preclinical therapies, the varying degree of impact on the mouse models, and potential measures of treatment effect are reviewed. One key issue discussed is the variable outcome of increasing SMN at different stages of disease progression.
Copyright © 2013 American Neurological Association.
Figures
References
-
- Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995 Jan 13;80(1):155–65. - PubMed
-
- Prior TW, Snyder PJ, Rink BD, et al. Newborn and carrier screening for spinal muscular atrophy. Am J Med Genet A. 2010 Jul;152A(7):1608–16. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
