

Transcriptional blood signatures distinguish pulmonary tuberculosis, pulmonary sarcoidosis, pneumonias and lung cancers
- PMID: 23940611
- PMCID: PMC3734176
- DOI: 10.1371/journal.pone.0070630
Transcriptional blood signatures distinguish pulmonary tuberculosis, pulmonary sarcoidosis, pneumonias and lung cancers
Erratum in
- PLoS One. 2013;8(8).doi: 10.1371/annotation/7d9ec449-aee0-48fe-8111-0c110850c0c1. Dominique, Valeyre [corrected to Valeyre, Dominique]
Abstract
Rationale: New approaches to define factors underlying the immunopathogenesis of pulmonary diseases including sarcoidosis and tuberculosis are needed to develop new treatments and biomarkers. Comparing the blood transcriptional response of tuberculosis to other similar pulmonary diseases will advance knowledge of disease pathways and help distinguish diseases with similar clinical presentations.
Objectives: To determine the factors underlying the immunopathogenesis of the granulomatous diseases, sarcoidosis and tuberculosis, by comparing the blood transcriptional responses in these and other pulmonary diseases.
Methods: We compared whole blood genome-wide transcriptional profiles in pulmonary sarcoidosis, pulmonary tuberculosis, to community acquired pneumonia and primary lung cancer and healthy controls, before and after treatment, and in purified leucocyte populations.
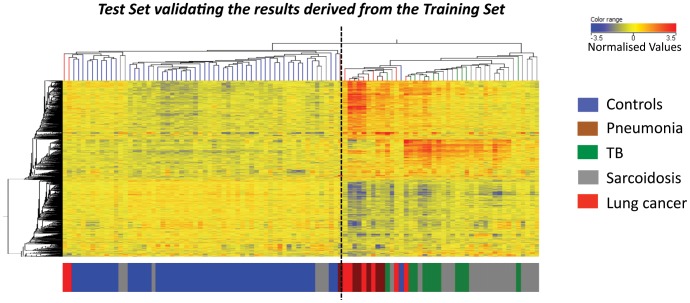
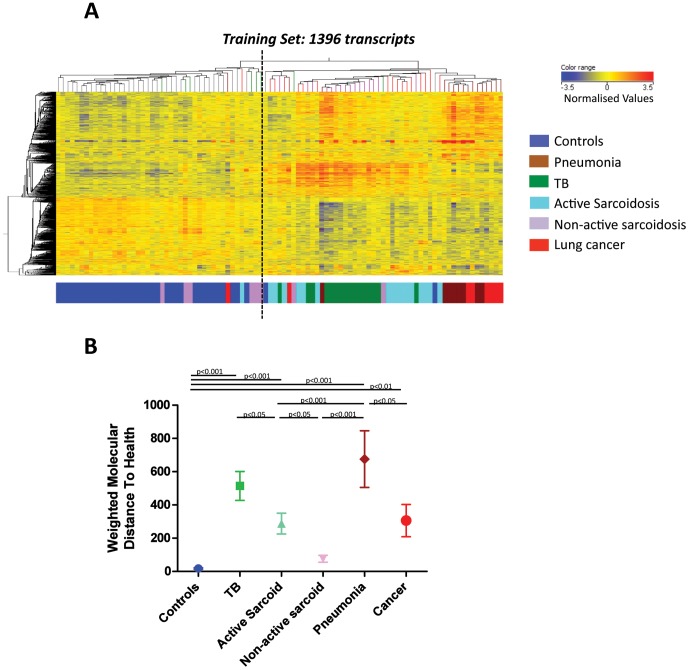
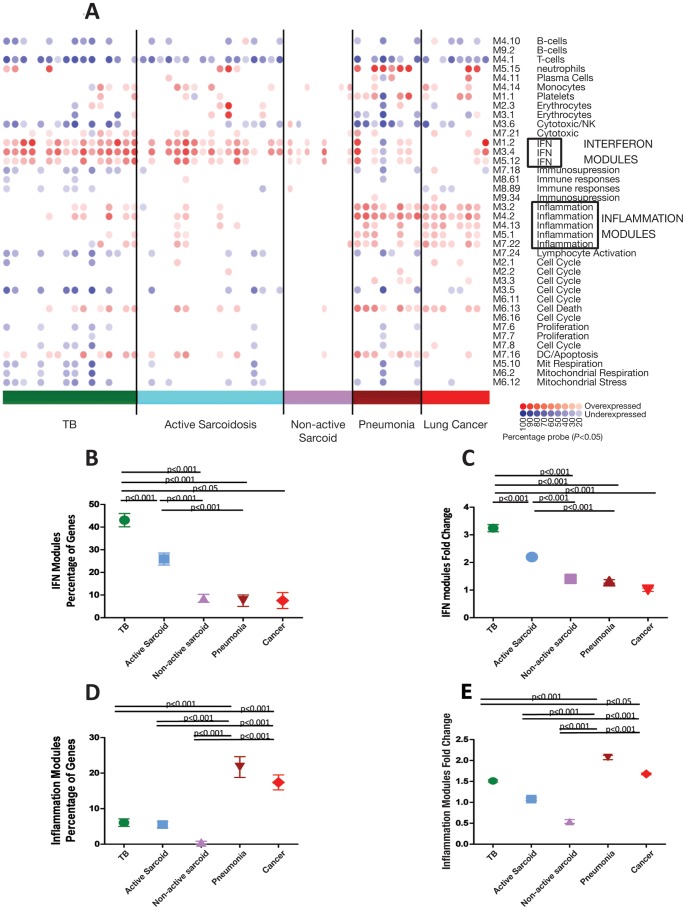
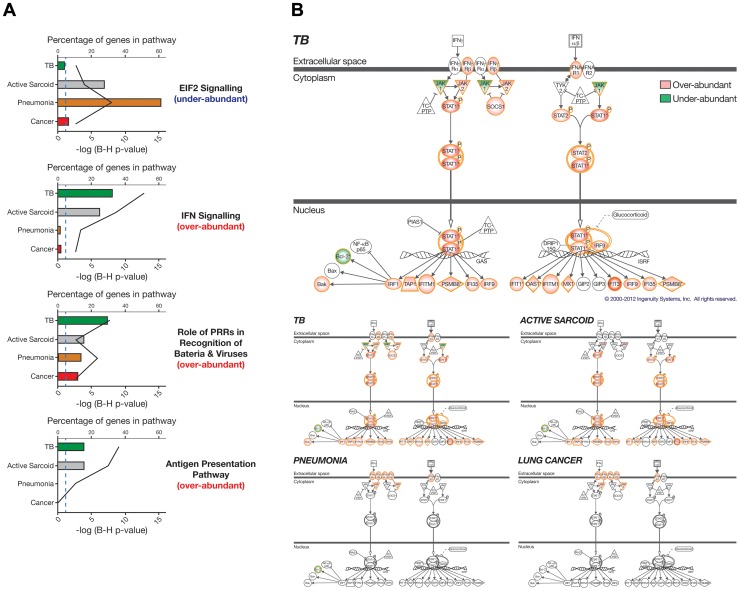
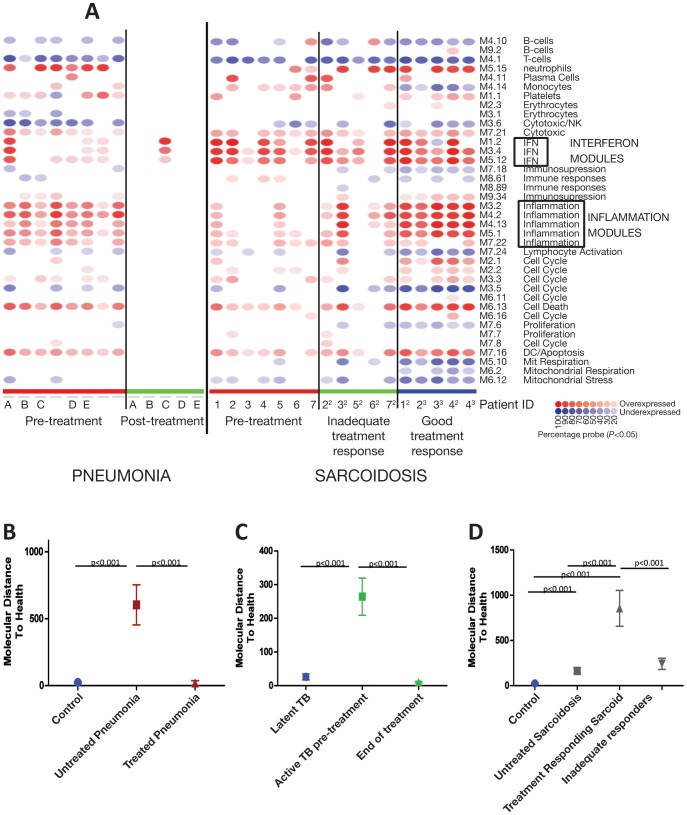
Measurements and main results: An Interferon-inducible neutrophil-driven blood transcriptional signature was present in both sarcoidosis and tuberculosis, with a higher abundance and expression in tuberculosis. Heterogeneity of the sarcoidosis signature correlated significantly with disease activity. Transcriptional profiles in pneumonia and lung cancer revealed an over-abundance of inflammatory transcripts. After successful treatment the transcriptional activity in tuberculosis and pneumonia patients was significantly reduced. However the glucocorticoid-responsive sarcoidosis patients showed a significant increase in transcriptional activity. 144-blood transcripts were able to distinguish tuberculosis from other lung diseases and controls.
Conclusions: Tuberculosis and sarcoidosis revealed similar blood transcriptional profiles, dominated by interferon-inducible transcripts, while pneumonia and lung cancer showed distinct signatures, dominated by inflammatory genes. There were also significant differences between tuberculosis and sarcoidosis in the degree of their transcriptional activity, the heterogeneity of their profiles and their transcriptional response to treatment.
Conflict of interest statement
Figures


References
-
- WHO (2010) Global tuberculosis control. World Health Organisation.
-
- Newman LS, Rose CS, Bresnitz EA, Rossman MD, Barnard J, et al. (2004) A case control etiologic study of sarcoidosis: environmental and occupational risk factors. Am J Respir Crit Care Med 170: 1324–1330. - PubMed
-
- Cliff JM, Lee JS, Constantinou N, Cho JE, Clark TG, et al. (2012) Distinct Phases of Blood Gene Expression Pattern Through Tuberculosis Treatment Reflect Modulation of the Humoral Immune Response. J Infect Dis. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
