

Genetics of amyotrophic lateral sclerosis: an update
- PMID: 23941283
- PMCID: PMC3766231
- DOI: 10.1186/1750-1326-8-28
Genetics of amyotrophic lateral sclerosis: an update
Abstract
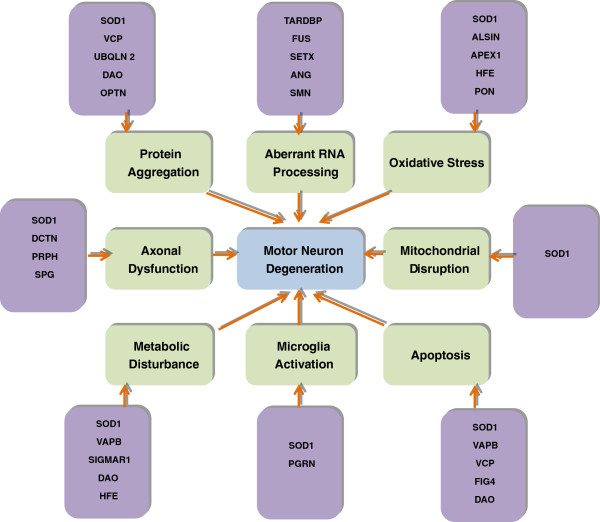
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder involving both upper motor neurons (UMN) and lower motor neurons (LMN). Enormous research has been done in the past few decades in unveiling the genetics of ALS, successfully identifying at least fifteen candidate genes associated with familial and sporadic ALS. Numerous studies attempting to define the pathogenesis of ALS have identified several plausible determinants and molecular pathways leading to motor neuron degeneration, which include oxidative stress, glutamate excitotoxicity, apoptosis, abnormal neurofilament function, protein misfolding and subsequent aggregation, impairment of RNA processing, defects in axonal transport, changes in endosomal trafficking, increased inflammation, and mitochondrial dysfunction. This review is to update the recent discoveries in genetics of ALS, which may provide insight information to help us better understanding of the disease neuropathogenesis.
Figures
References
-
- Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7:710–723. - PubMed
-
- Czaplinski A, Yen AA, Simpson EP, Appel SH. Slower disease progression and prolonged survival in contemporary patients with amyotrophic lateral sclerosis: is the natural history of amyotrophic lateral sclerosis changing? Arch Neurol. 2006;63:1139–1143. doi: 10.1001/archneur.63.8.1139. - DOI - PubMed
-
- Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, Kinoshita Y, Kamada M, Nodera H, Suzuki H, Komure O, Matsuura S, Kobatake K, Morimoto N, Abe K, Suzuki N, Aoki M, Kawata A, Hirai T, Kato T, Ogasawara K, Hirano A, Takumi T, Kusaka H, Hagiwara K, Kaji R, Kawakami H. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465:223–226. doi: 10.1038/nature08971. - DOI - PubMed
-
- Turner MR, Hardiman O, Benatar M, Brooks BR, Chio A, de Carvalho M, Ince PG, Lin C, Miller RG, Mitsumoto H, Nicholson G, Ravits J, Shaw PJ, Swash M, Talbot K, Traynor BJ, Van den Berg LH, Veldink JH, Vucic S, Kiernan MC. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 2013;12:310–322. doi: 10.1016/S1474-4422(13)70036-X. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
