

PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes
- PMID: 23942190
- PMCID: PMC3760377
- DOI: 10.1038/ncomms3304
PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes
Abstract
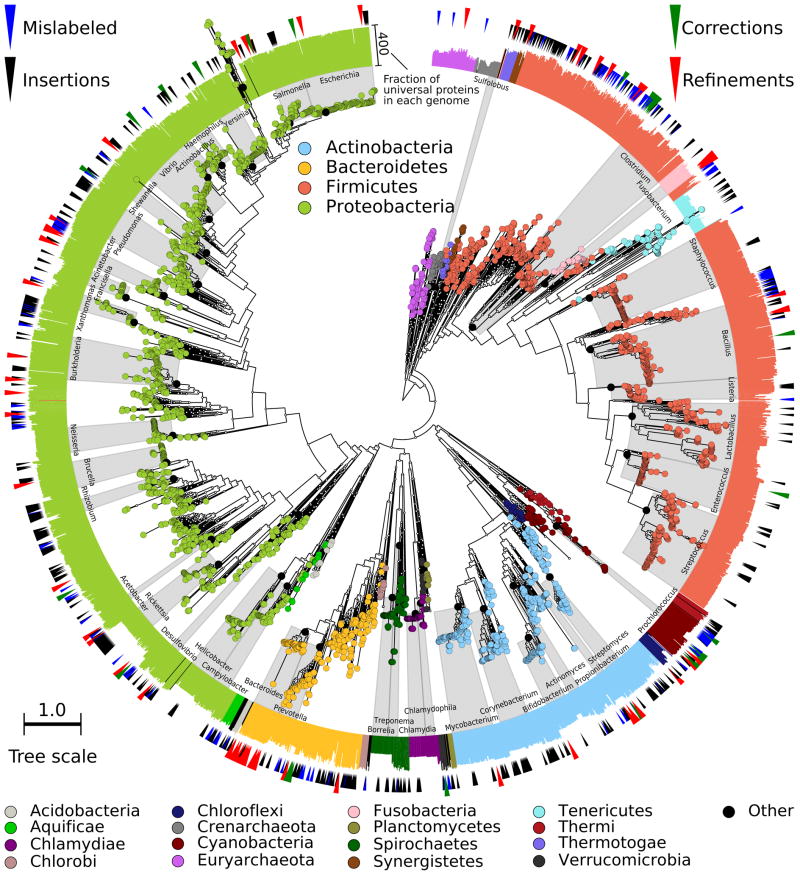
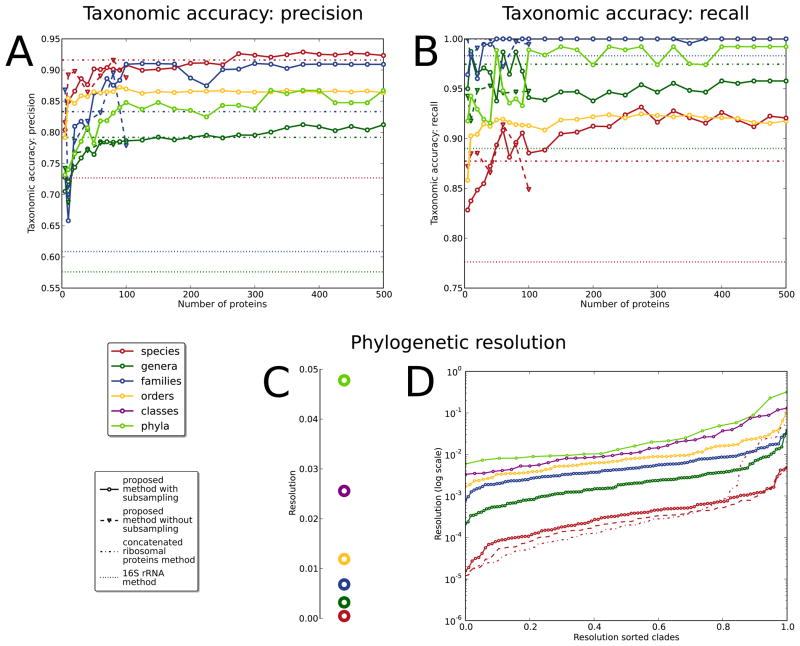
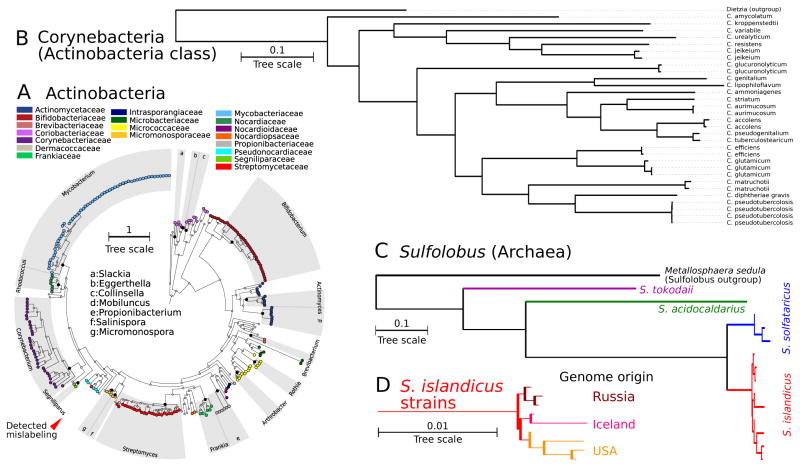
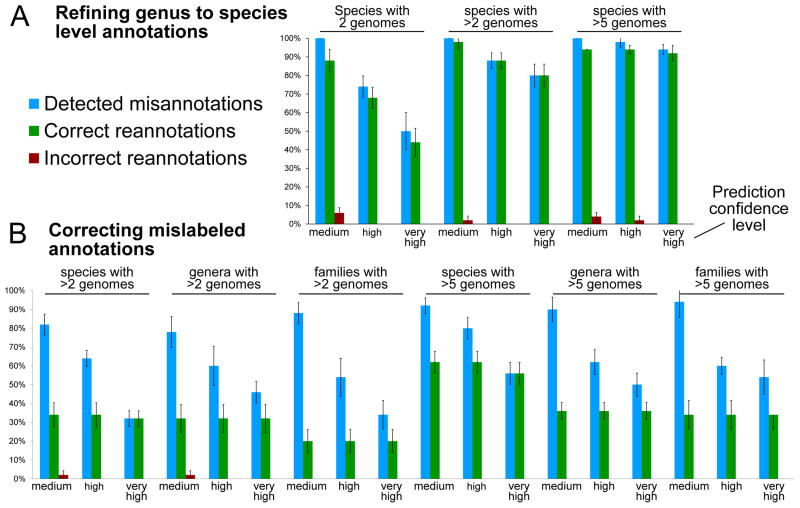
New microbial genomes are constantly being sequenced, and it is crucial to accurately determine their taxonomic identities and evolutionary relationships. Here we report PhyloPhlAn, a new method to assign microbial phylogeny and putative taxonomy using >400 proteins optimized from among 3,737 genomes. This method measures the sequence diversity of all clades, classifies genomes from deep-branching candidate divisions through closely related subspecies and improves consistency between phylogenetic and taxonomic groupings. PhyloPhlAn improved taxonomic accuracy for existing and newly sequenced genomes, detecting 157 erroneous labels, correcting 46 and placing or refining 130 new genomes. We provide examples of accurate classifications from subspecies (Sulfolobus spp.) to phyla, and of preliminary rooting of deep-branching candidate divisions, including consistent statistical support for Caldiserica (formerly candidate division OP5). PhyloPhlAn will thus be useful for both phylogenetic assessment and taxonomic quality control of newly sequenced genomes. The final phylogenies, conserved protein sequences and open-source implementation are available online.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Ochman H, Wilson Evolution in bacteria: evidence for a universal substitution rate in cellular genomes. J Mol Evol. 1987;26:74. - PubMed
-
- Gogarten JP, Townsend JP. Horizontal gene transfer, genome innovation and evolution. Nat Rev Microbiol. 2005;3:679–687. - PubMed
-
- Gardy JL, et al. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. New Engl J Med. 2011;364:730. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
