

The brain reward circuitry in mood disorders
- PMID: 23942470
- PMCID: PMC3867253
- DOI: 10.1038/nrn3381
The brain reward circuitry in mood disorders
Erratum in
- Nat Rev Neurosci. 2013 Oct;14(10):736
Abstract
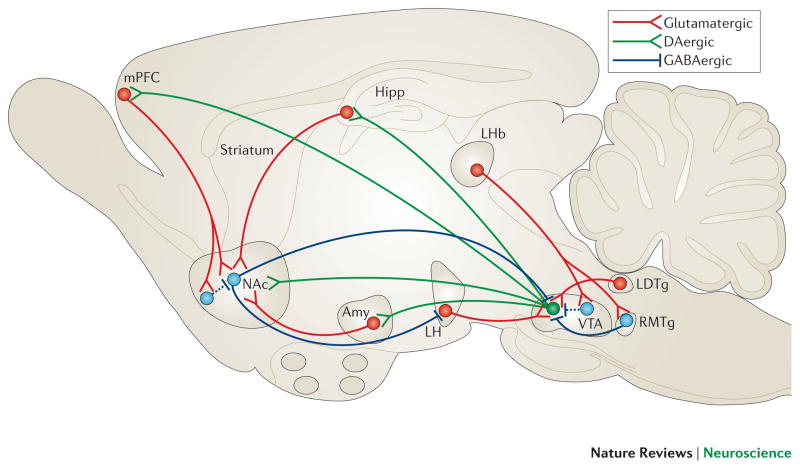
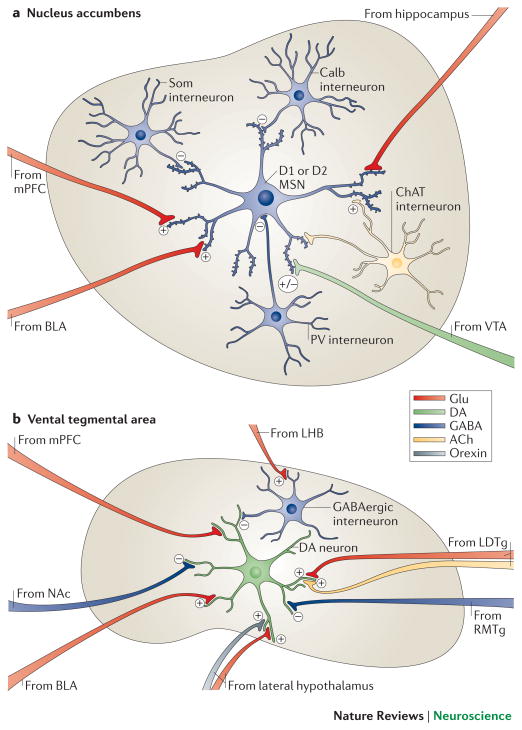
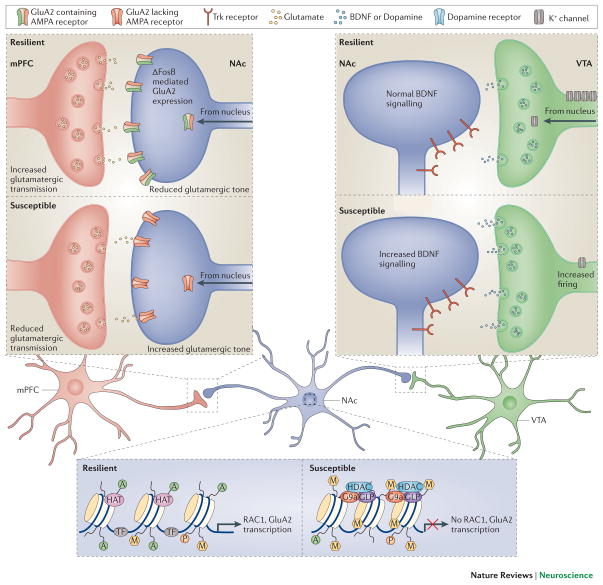
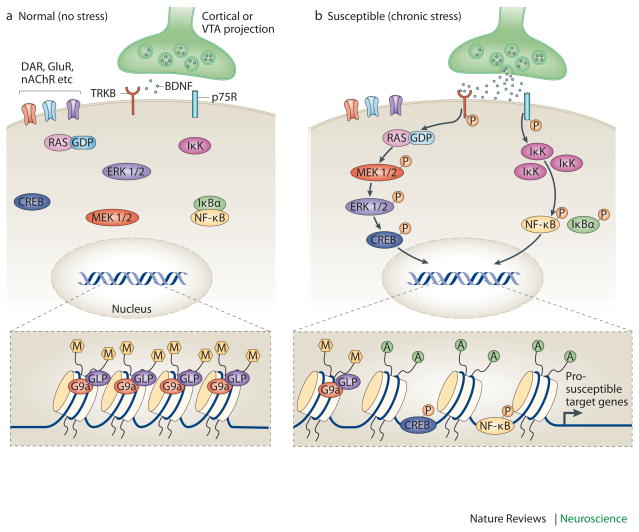
Mood disorders are common and debilitating conditions characterized in part by profound deficits in reward-related behavioural domains. A recent literature has identified important structural and functional alterations within the brain's reward circuitry--particularly in the ventral tegmental area-nucleus accumbens pathway--that are associated with symptoms such as anhedonia and aberrant reward-associated perception and memory. This Review synthesizes recent data from human and rodent studies from which emerges a circuit-level framework for understanding reward deficits in depression. We also discuss some of the molecular and cellular underpinnings of this framework, ranging from adaptations in glutamatergic synapses and neurotrophic factors to transcriptional and epigenetic mechanisms.
Figures
References
-
- Culpepper L. Why do you need to move beyond first-line therapy for major depression? J Clin Psychiatry. 71(Suppl 1):4–9. - PubMed
-
- Conway KP, Compton W, Stinson FS, Grant BF. Lifetime comorbidity of DSM-IV mood and anxiety disorders and specific drug use disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. J Clin Psychiatry. 2006;67:247–57. - PubMed
-
- Nestler EJ, Carlezon WA., Jr The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59:1151–9. - PubMed
-
- Koob GF, Le Moal M. Addiction and the brain antireward system. Annu Rev Psychol. 2008;59:29–53. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
