

BCL-2 inhibition with ABT-737 prolongs survival in an NRAS/BCL-2 mouse model of AML by targeting primitive LSK and progenitor cells
- PMID: 23943652
- PMCID: PMC3799000
- DOI: 10.1182/blood-2012-07-445635
BCL-2 inhibition with ABT-737 prolongs survival in an NRAS/BCL-2 mouse model of AML by targeting primitive LSK and progenitor cells
Abstract
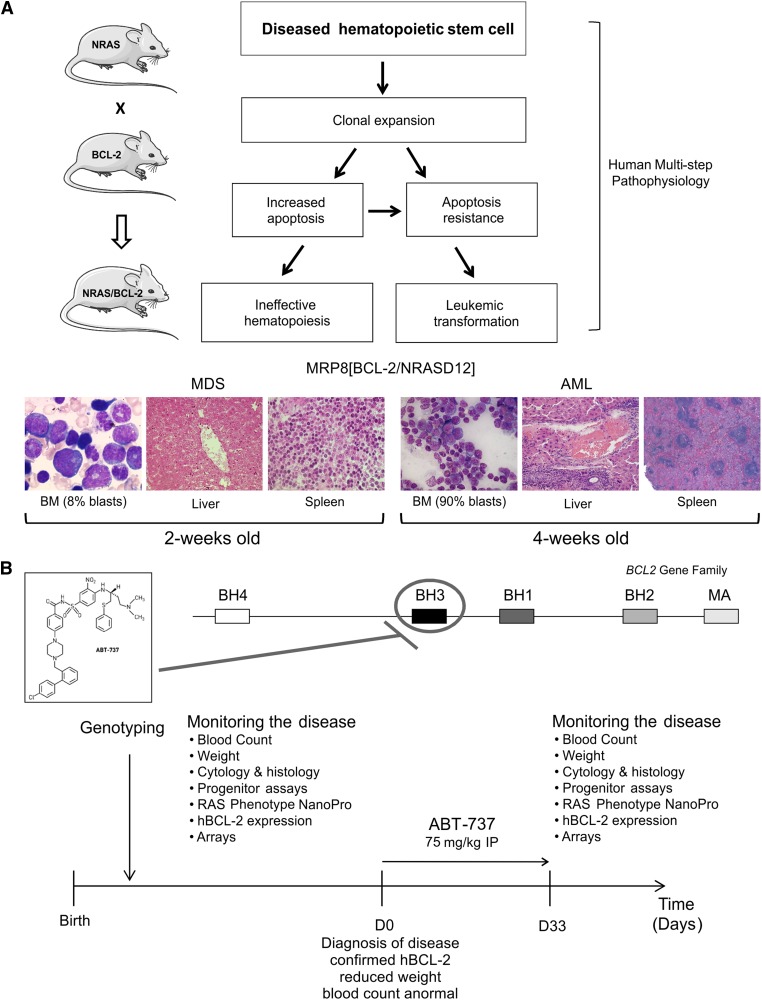
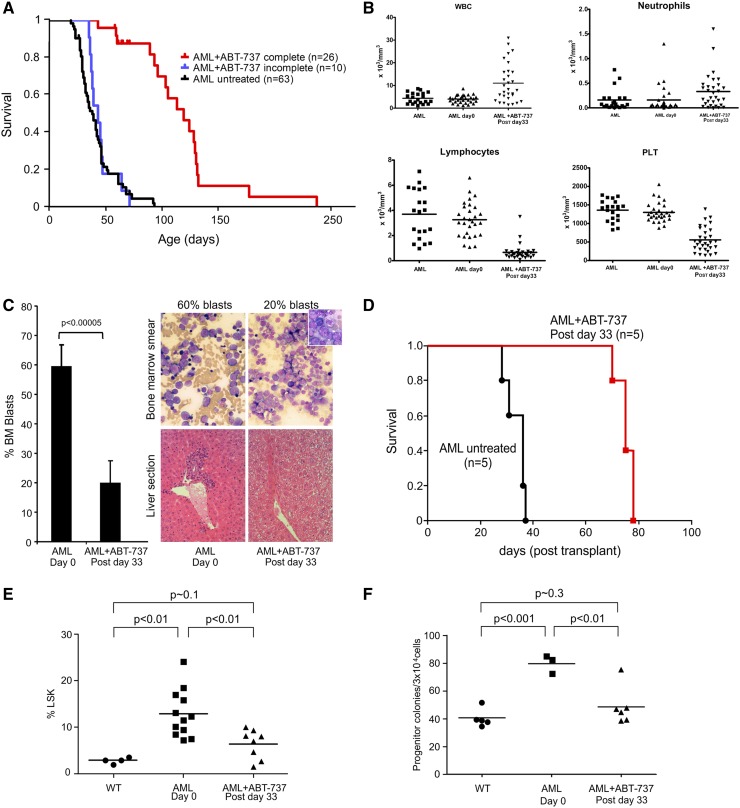
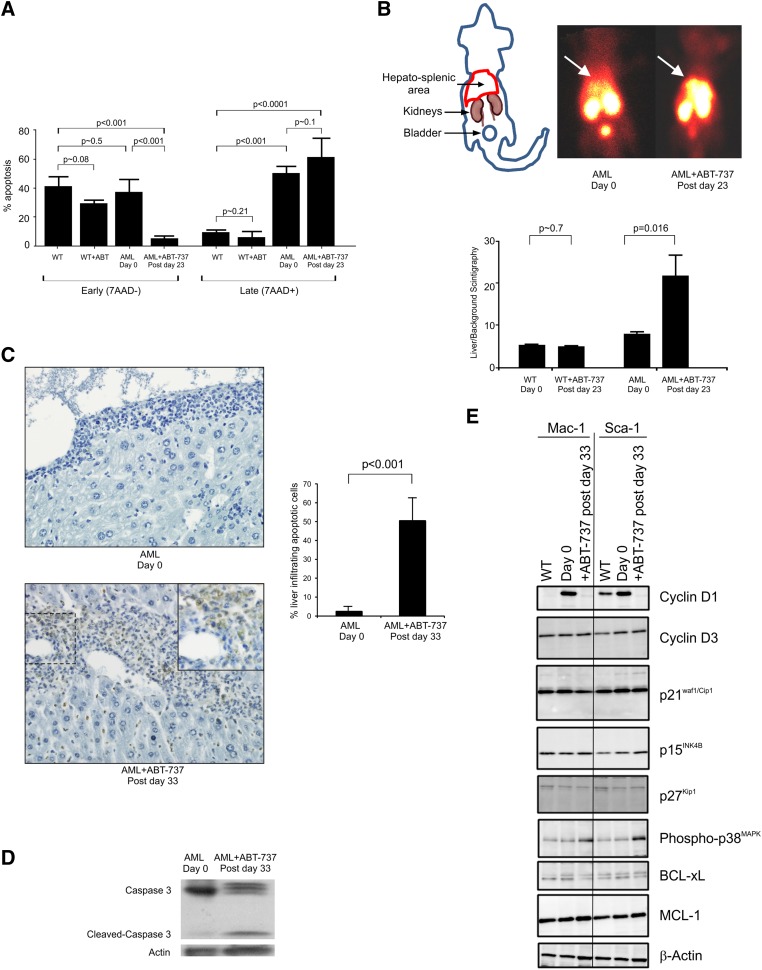
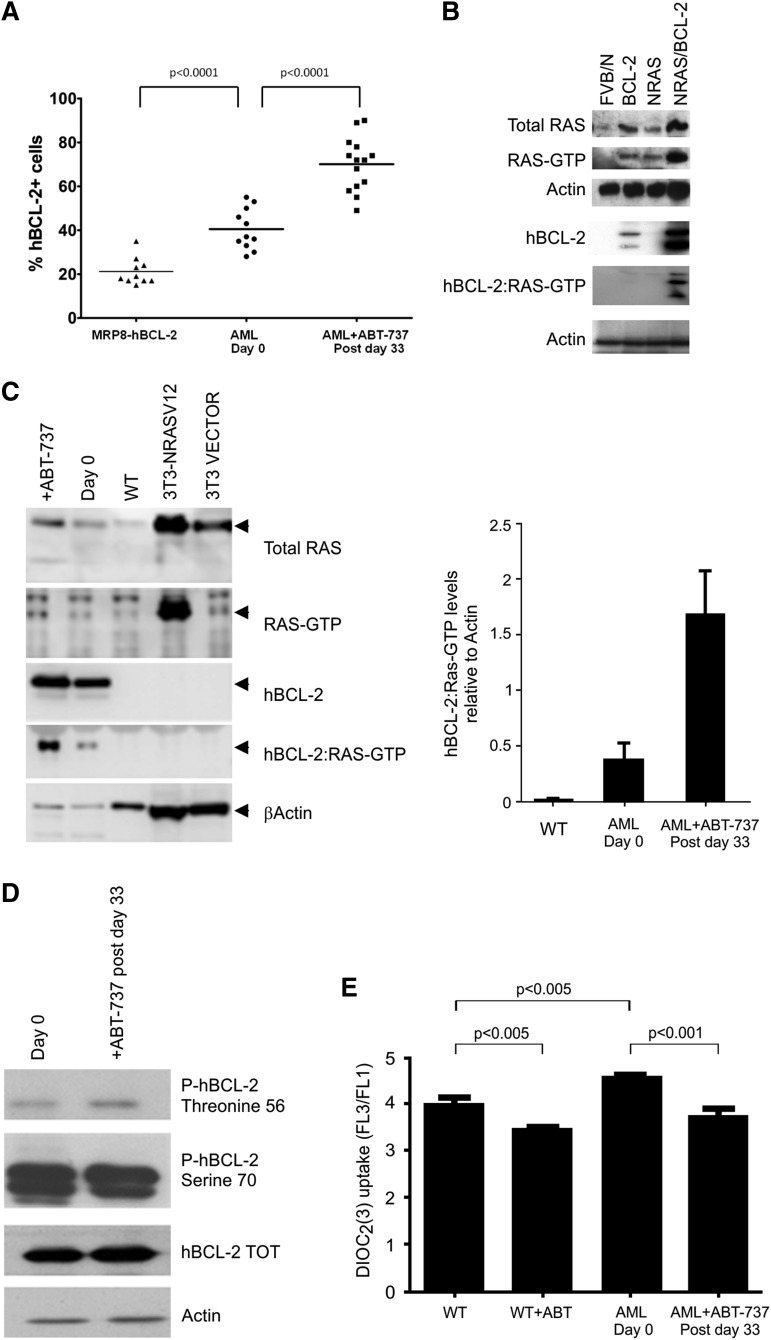
Myelodysplastic syndrome (MDS) transforms into an acute myelogenous leukemia (AML) with associated increased bone marrow (BM) blast infiltration. Using a transgenic mouse model, MRP8[NRASD12/hBCL-2], in which the NRAS:BCL-2 complex at the mitochondria induces MDS progressing to AML with dysplastic features, we studied the therapeutic potential of a BCL-2 homology domain 3 mimetic inhibitor, ABT-737. Treatment significantly extended lifespan, increased survival of lethally irradiated secondary recipients transplanted with cells from treated mice compared with cells from untreated mice, with a reduction of BM blasts, Lin-/Sca-1(+)/c-Kit(+), and progenitor populations by increased apoptosis of infiltrating blasts of diseased mice assessed in vivo by technicium-labeled annexin V single photon emission computed tomography and ex vivo by annexin V/7-amino actinomycin D flow cytometry, terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling, caspase 3 cleavage, and re-localization of the NRAS:BCL-2 complex from mitochondria to plasma membrane. Phosphoprotein analysis showed restoration of wild-type (WT) AKT or protein kinase B, extracellular signal-regulated kinase 1/2 and mitogen-activated protein kinase patterns in spleen cells after treatment, which showed reduced mitochondrial membrane potential. Exon specific gene expression profiling corroborates the reduction of leukemic cells, with an increase in expression of genes coding for stem cell development and maintenance, myeloid differentiation, and apoptosis. Myelodysplastic features persist underscoring targeting of BCL-2-mediated effects on MDS-AML transformation and survival of leukemic cells.
Figures
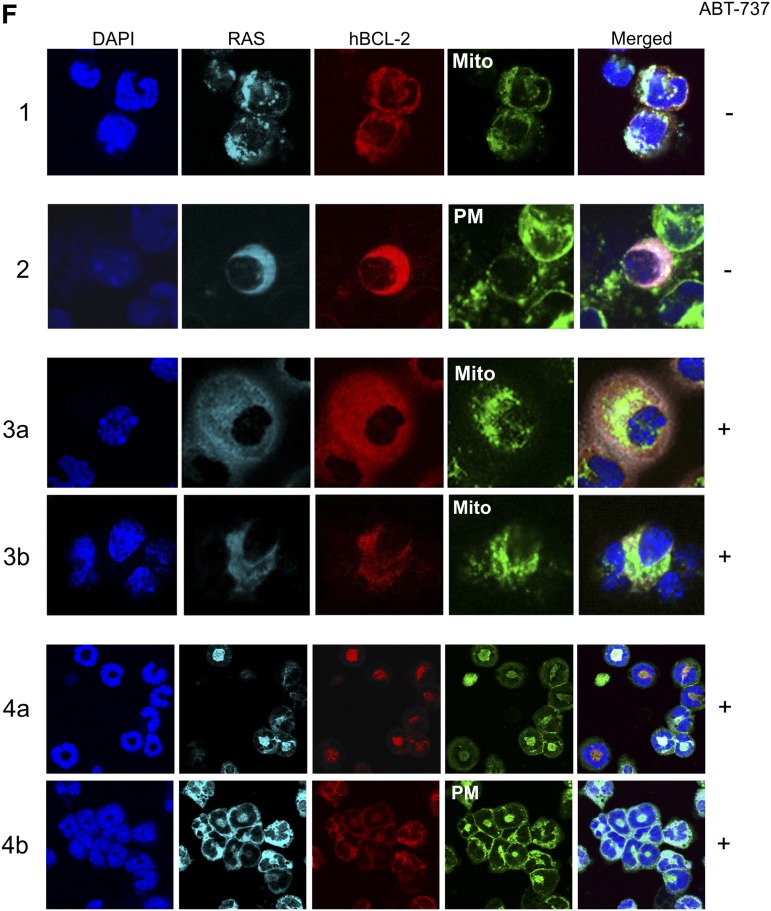
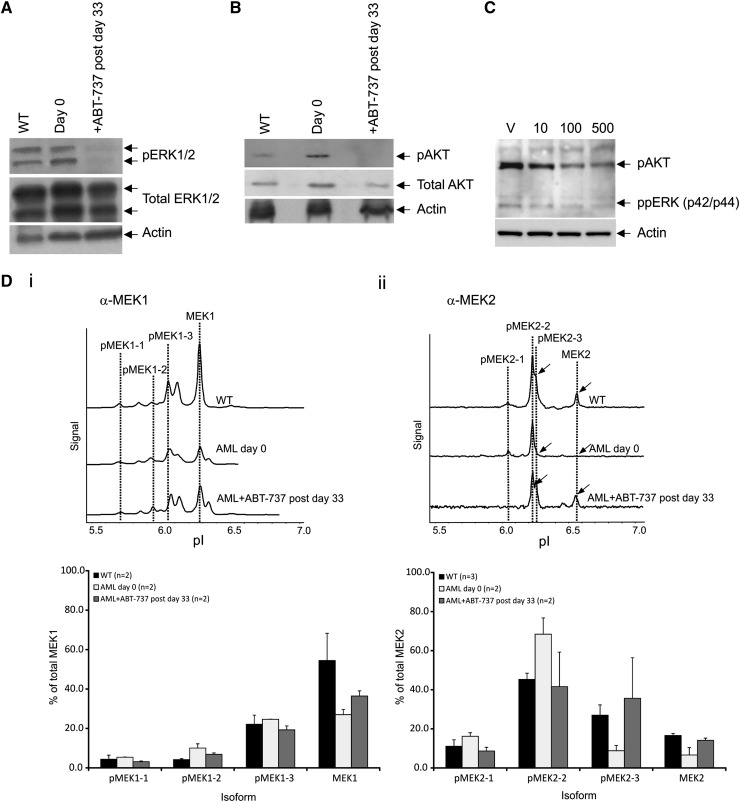
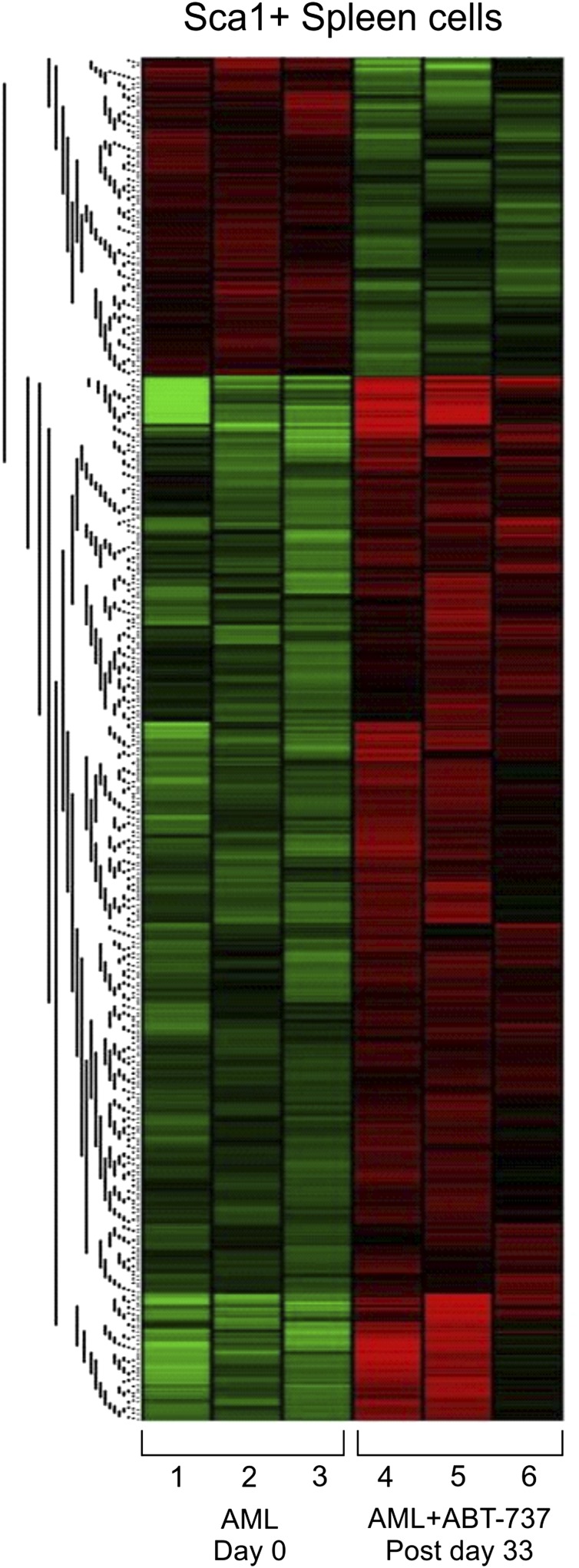
References
-
- Padua RA, Guinn BA, Al-Sabah AI, et al. RAS, FMS and p53 mutations and poor clinical outcome in myelodysplasias: a 10-year follow-up. Leukemia. 1998;12(6):887–892. - PubMed
-
- Omidvar N, Kogan S, Beurlet S, et al. BCL-2 and mutant NRAS interact physically and functionally in a mouse model of progressive myelodysplasia. Cancer Res. 2007;67(24):11657–11667. - PubMed
-
- Parker JE, Mufti GJ. The myelodysplastic syndromes: a matter of life or death. Acta Haematol. 2004;111(1-2):78–99. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
