

Sarm1-mediated axon degeneration requires both SAM and TIR interactions
- PMID: 23946415
- PMCID: PMC3742939
- DOI: 10.1523/JNEUROSCI.1197-13.2013
Sarm1-mediated axon degeneration requires both SAM and TIR interactions
Abstract
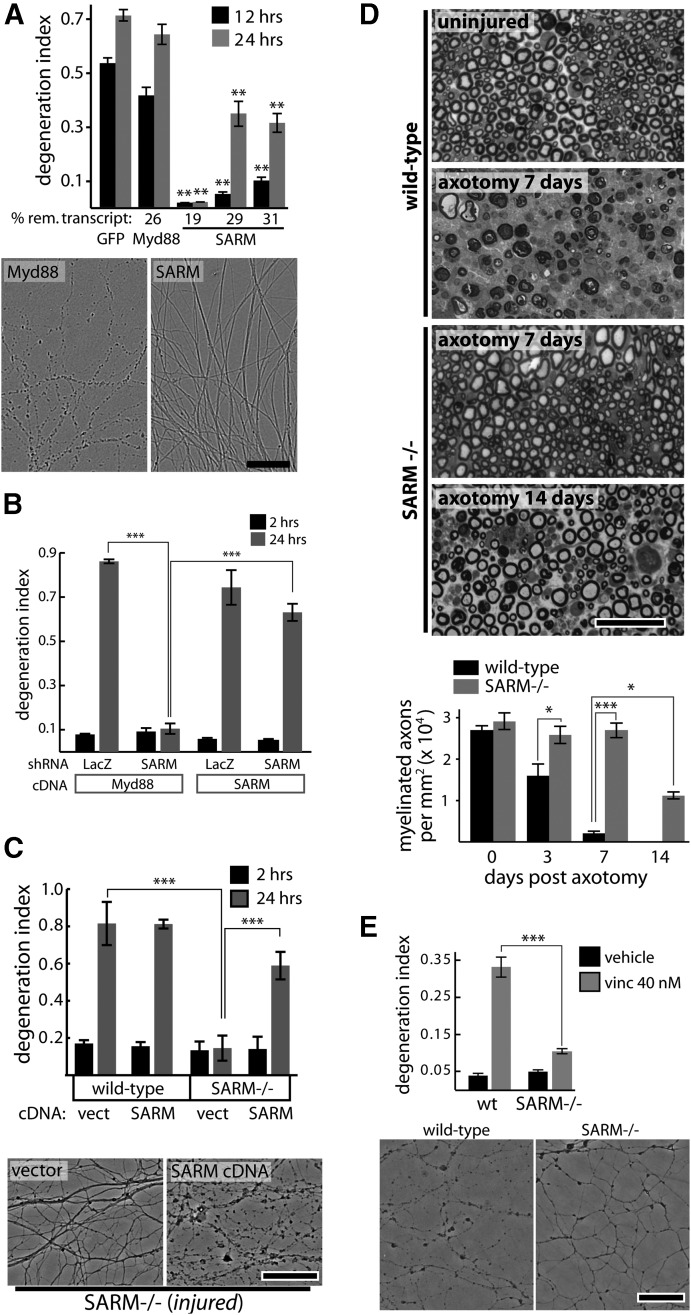
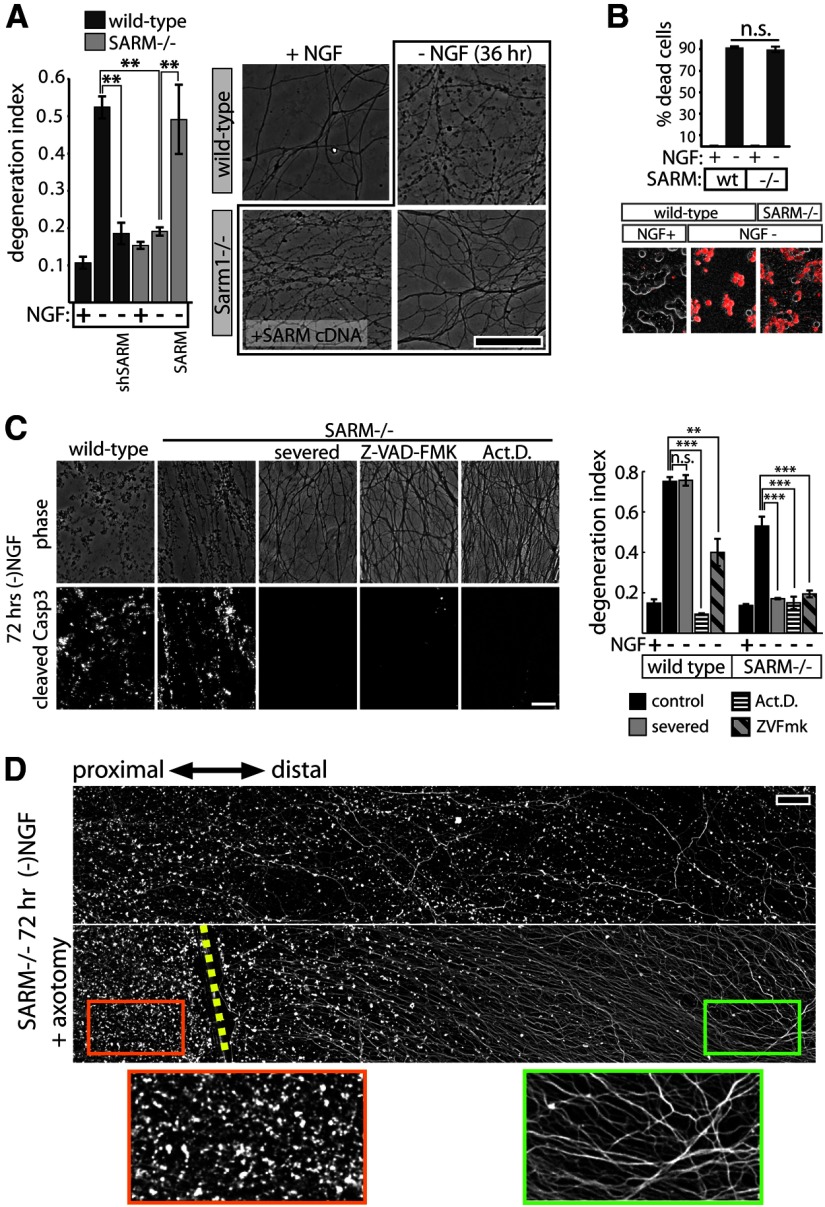
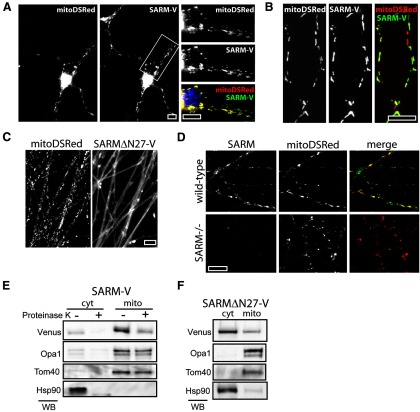
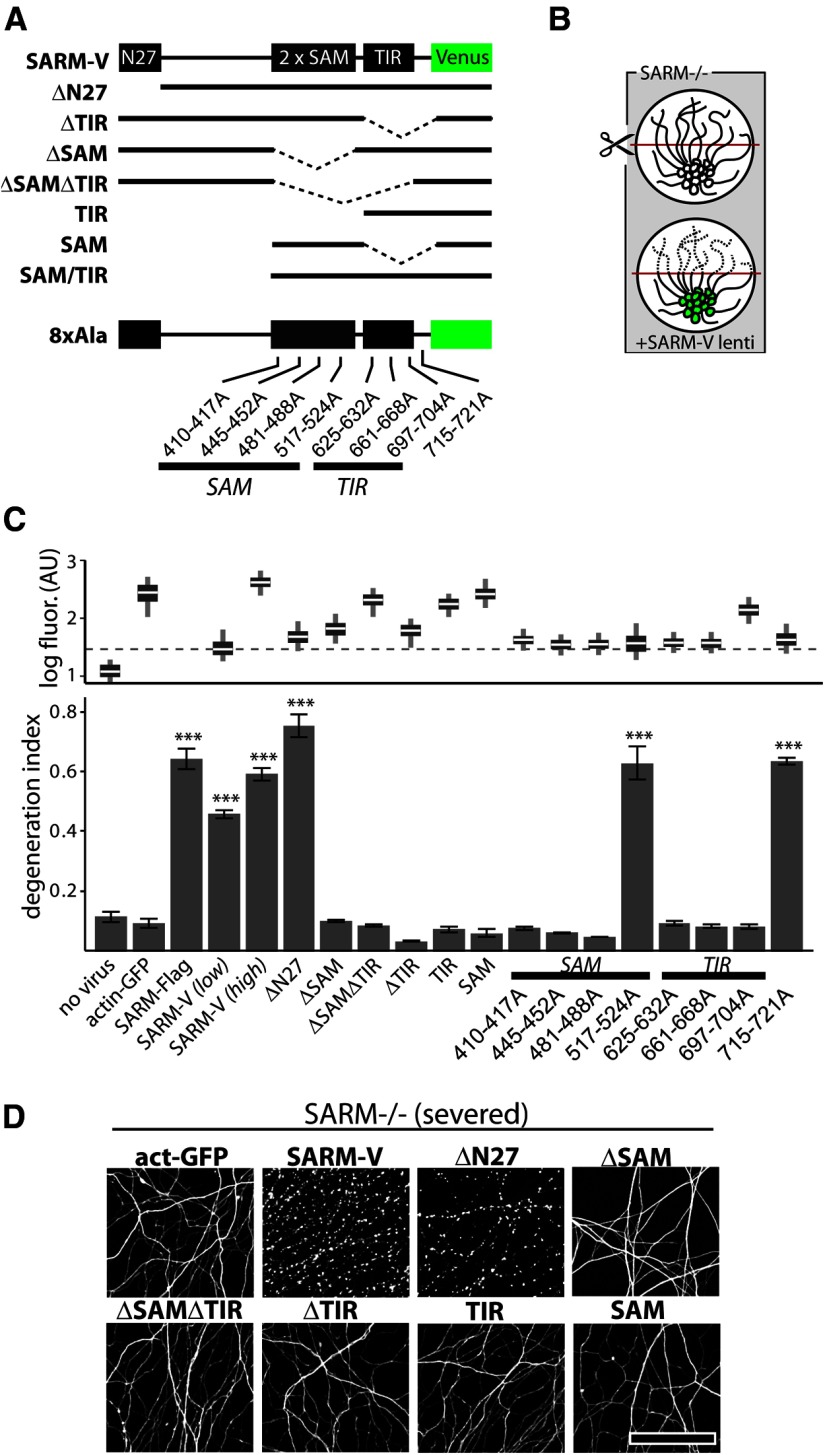
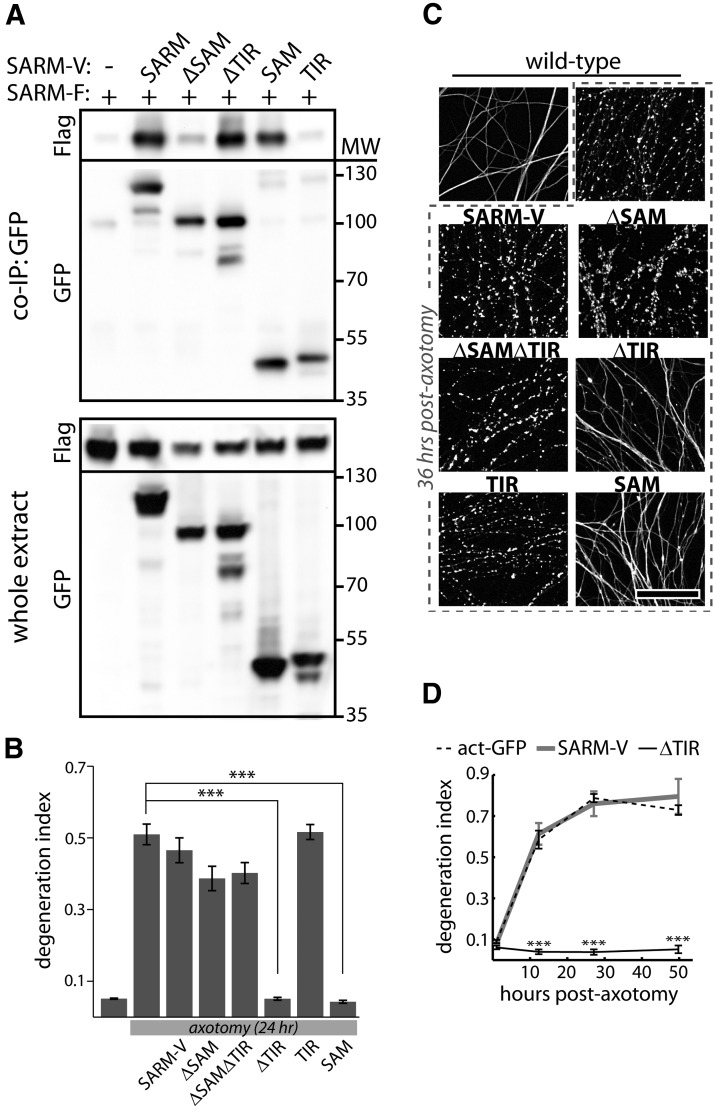
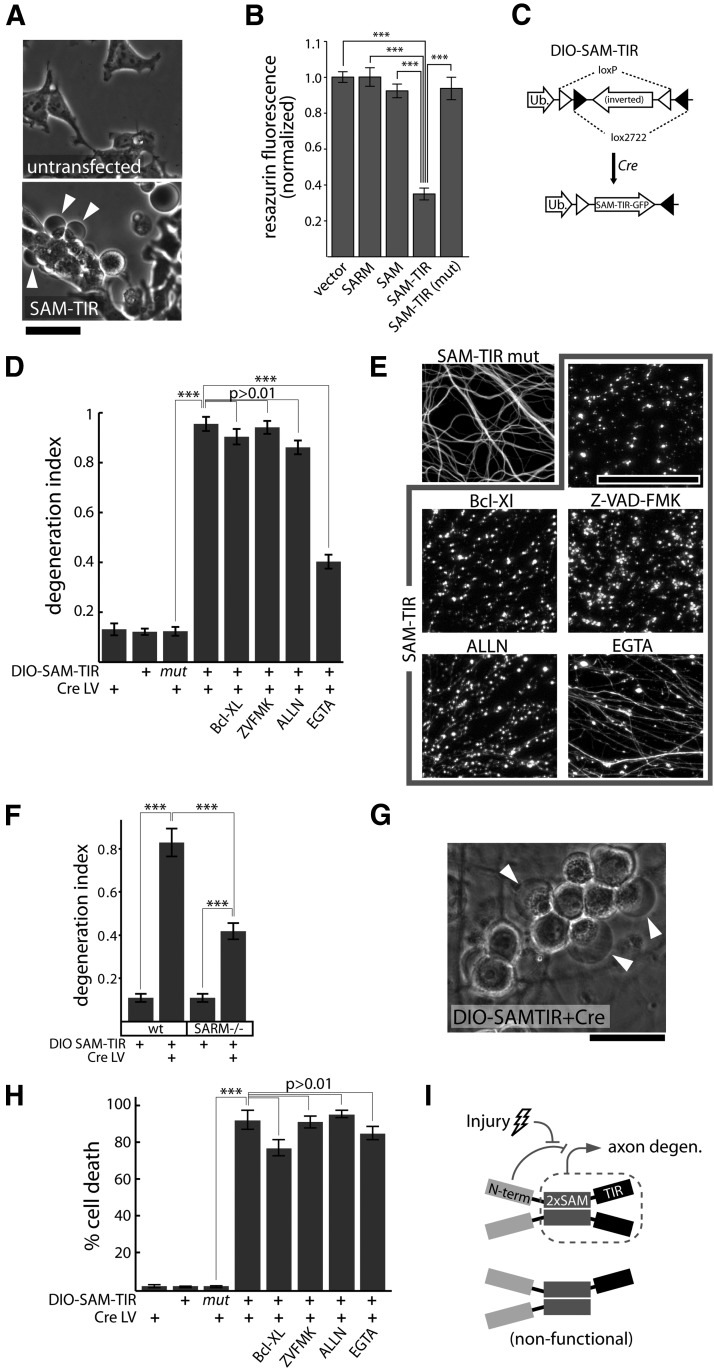
Axon degeneration is an evolutionarily conserved pathway that eliminates damaged or unneeded axons. Manipulation of this poorly understood pathway may allow treatment of a wide range of neurological disorders. In an RNAi-based screen performed in cultured mouse DRG neurons, we observed strong suppression of injury-induced axon degeneration upon knockdown of Sarm1 [SARM (sterile α-motif-containing and armadillo-motif containing protein)]. We find that a SARM-dependent degeneration program is engaged by disparate neuronal insults: SARM ablation blocks axon degeneration induced by axotomy or vincristine treatment, while SARM acts in parallel with a soma-derived caspase-dependent pathway following trophic withdrawal. SARM is a multidomain protein that associates with neuronal mitochondria. Deletion of the N-terminal mitochondrial localization sequence disrupts SARM mitochondrial localization in neurons but does not alter its ability to promote axon degeneration. In contrast, mutation of either the SAM (sterile α motif) or TIR (Toll-interleukin-1 receptor) domains abolishes the ability of SARM to promote axonal degeneration, while a SARM mutant containing only these domains elicits axon degeneration and nonapoptotic neuronal death even in the absence of injury. Protein-protein interaction studies demonstrate that the SAM domains are necessary and sufficient to mediate SARM-SARM binding. SARM mutants lacking a TIR domain bind full-length SARM and exhibit strong dominant-negative activity. These results indicate that SARM plays an integral role in the dismantling of injured axons and support a model in which SAM-mediated multimerization is necessary for TIR-dependent engagement of a downstream destruction pathway. These findings suggest that inhibitors of SAM and TIR interactions represent therapeutic candidates for blocking pathological axon loss and neuronal cell death.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials