

Amplification of distant estrogen response elements deregulates target genes associated with tamoxifen resistance in breast cancer
- PMID: 23948299
- PMCID: PMC3890247
- DOI: 10.1016/j.ccr.2013.07.007
Amplification of distant estrogen response elements deregulates target genes associated with tamoxifen resistance in breast cancer
Abstract
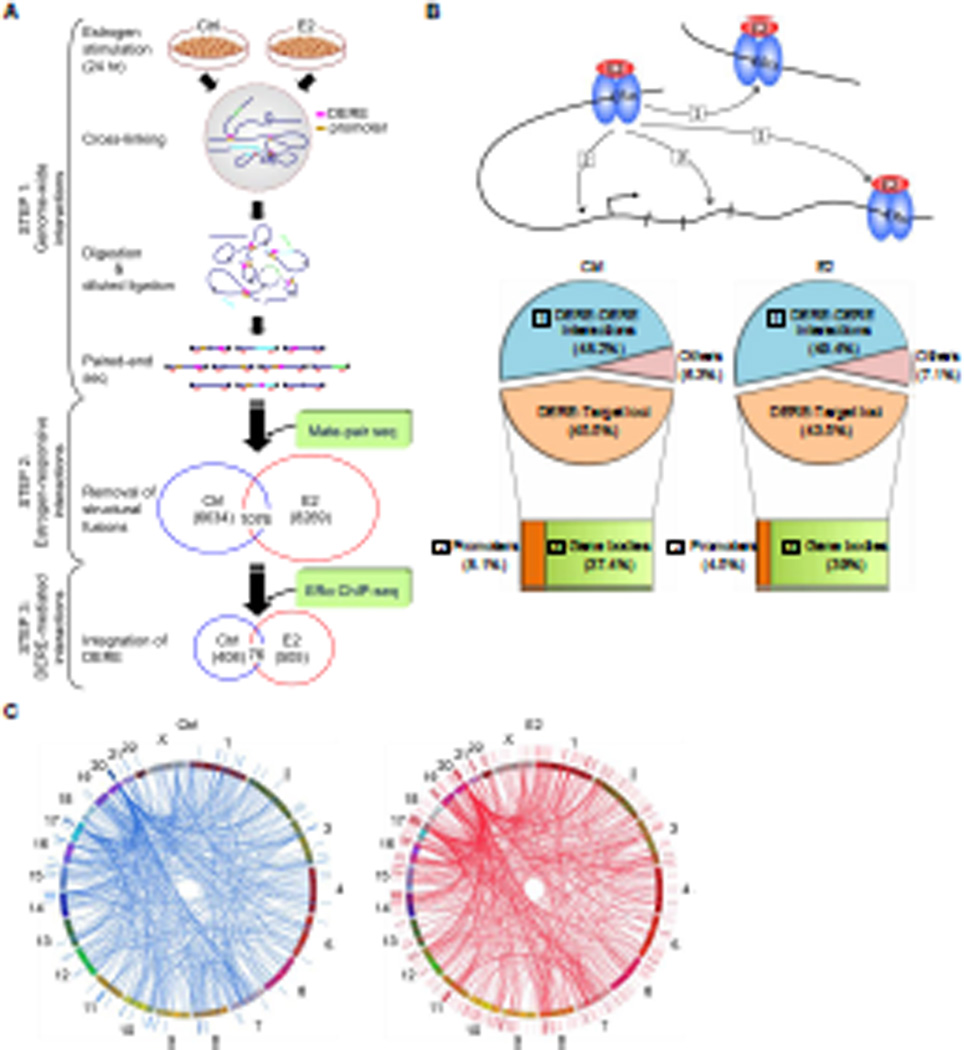
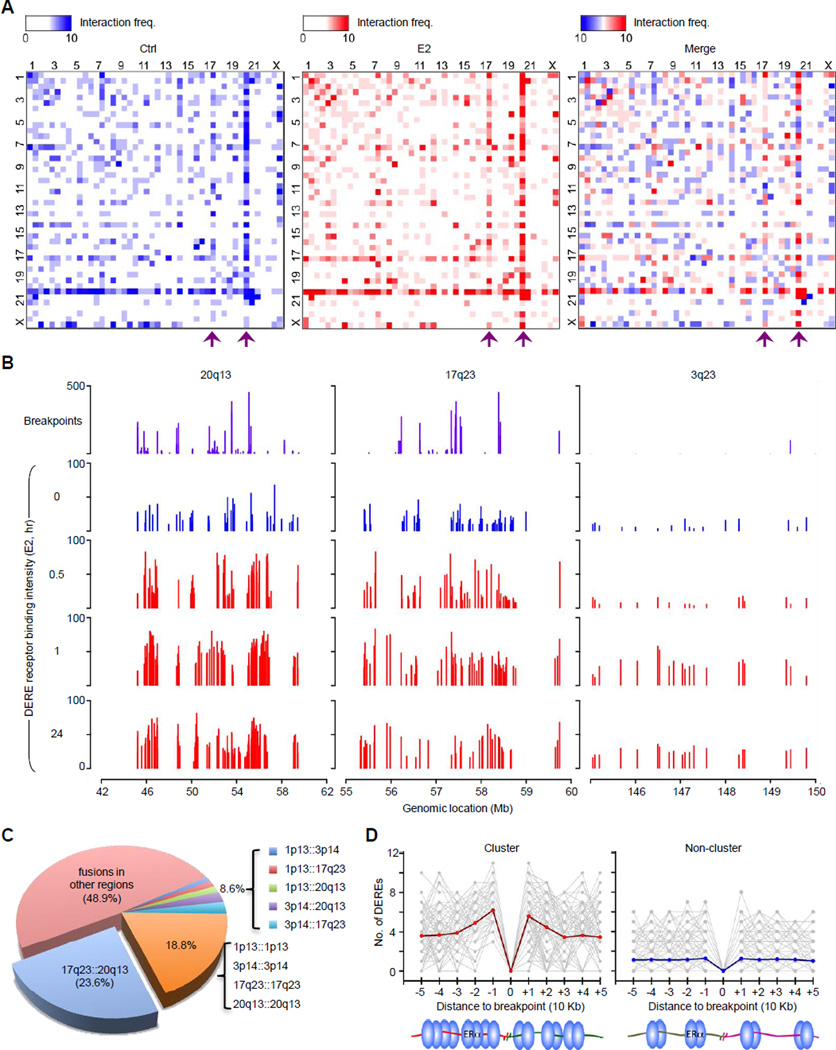
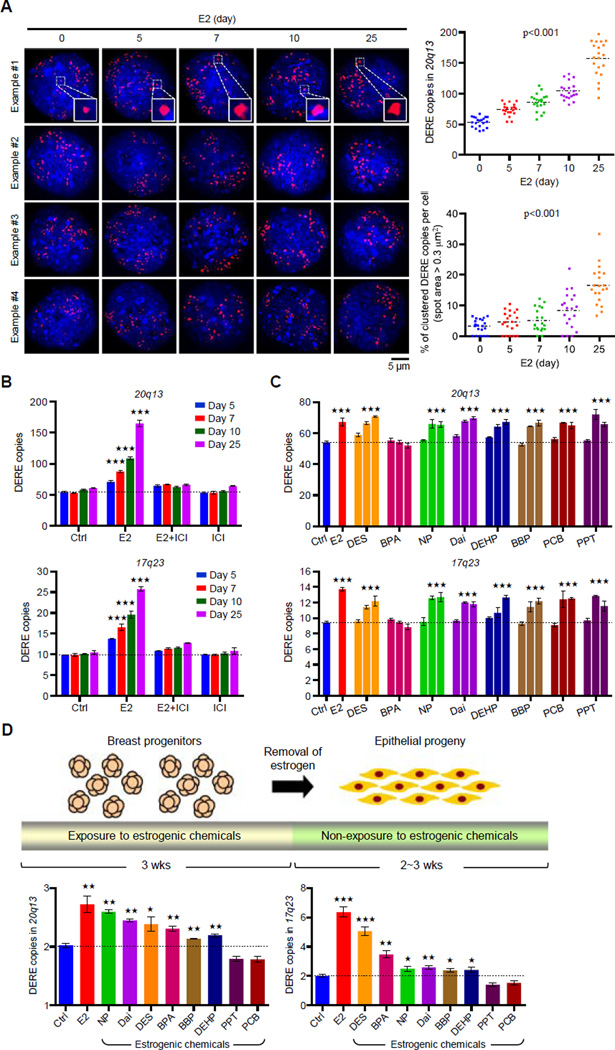
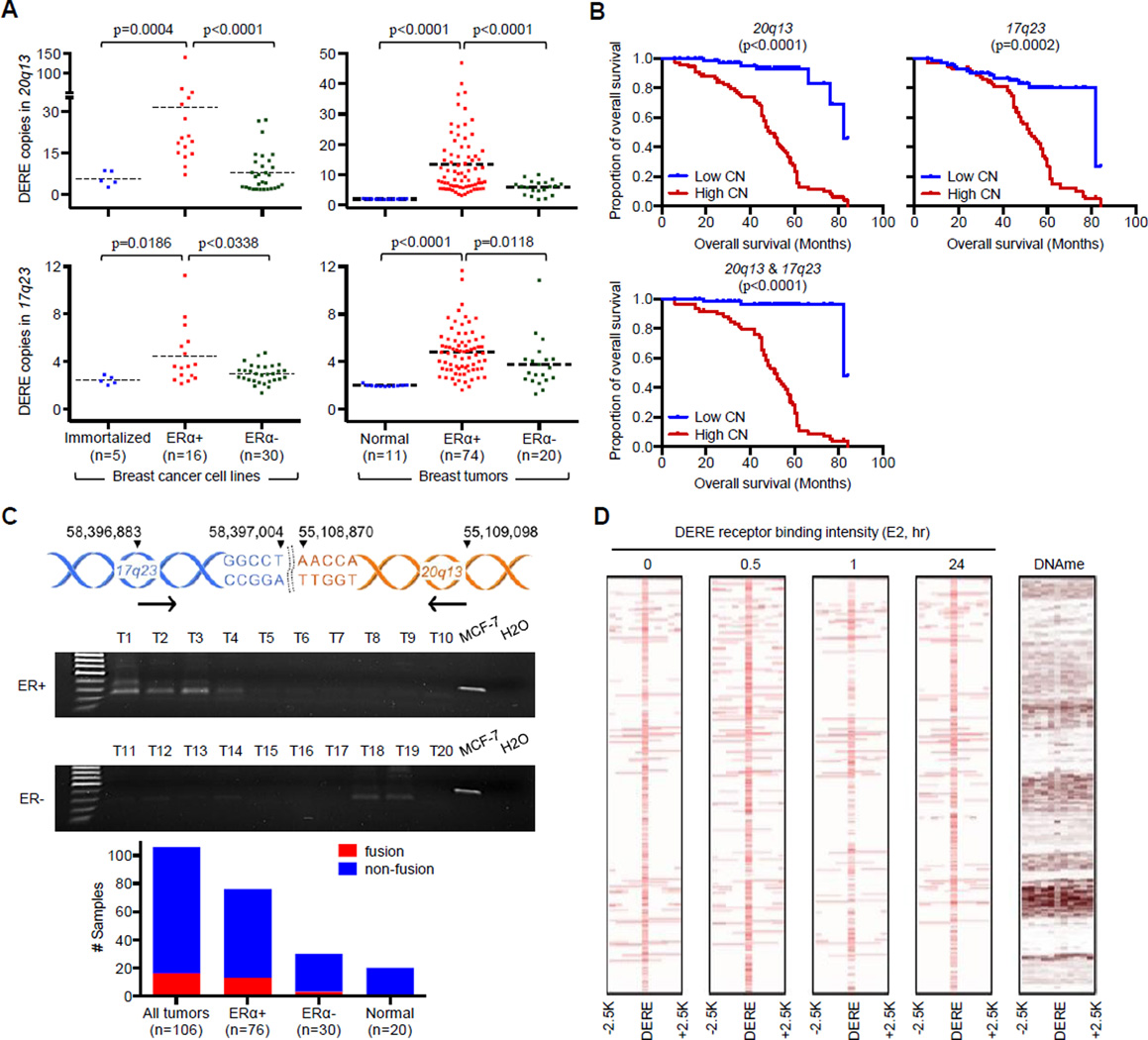
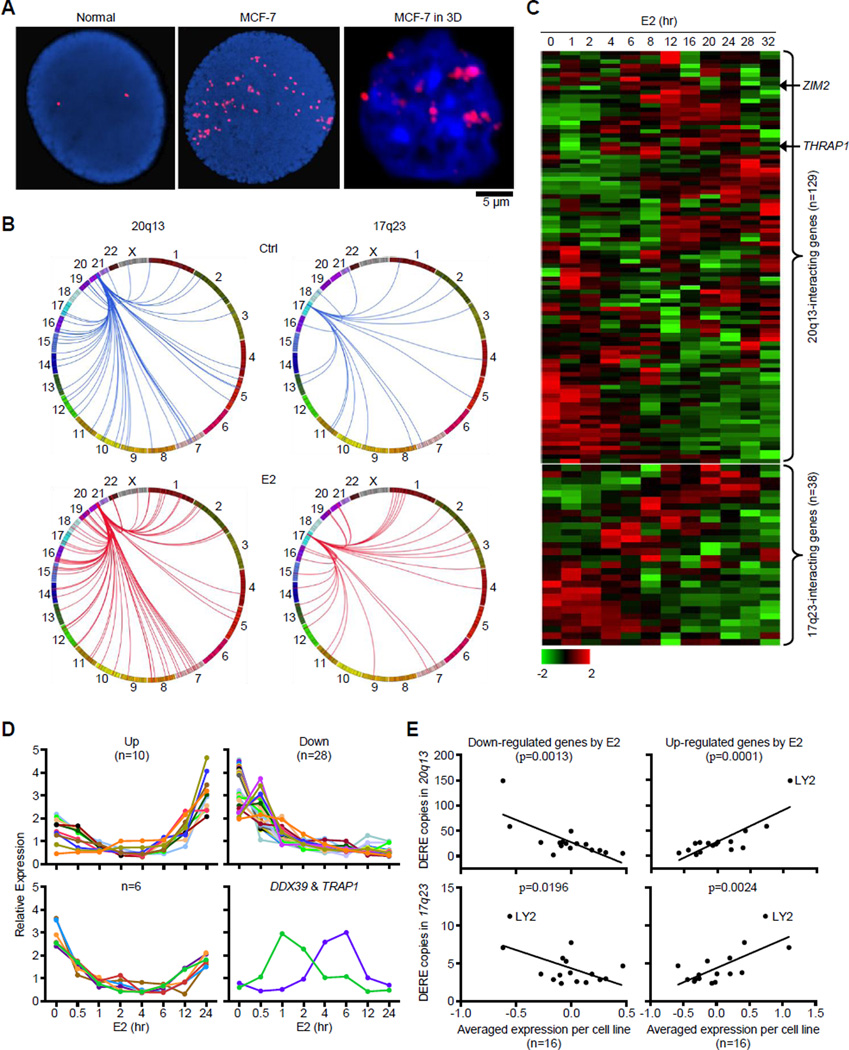
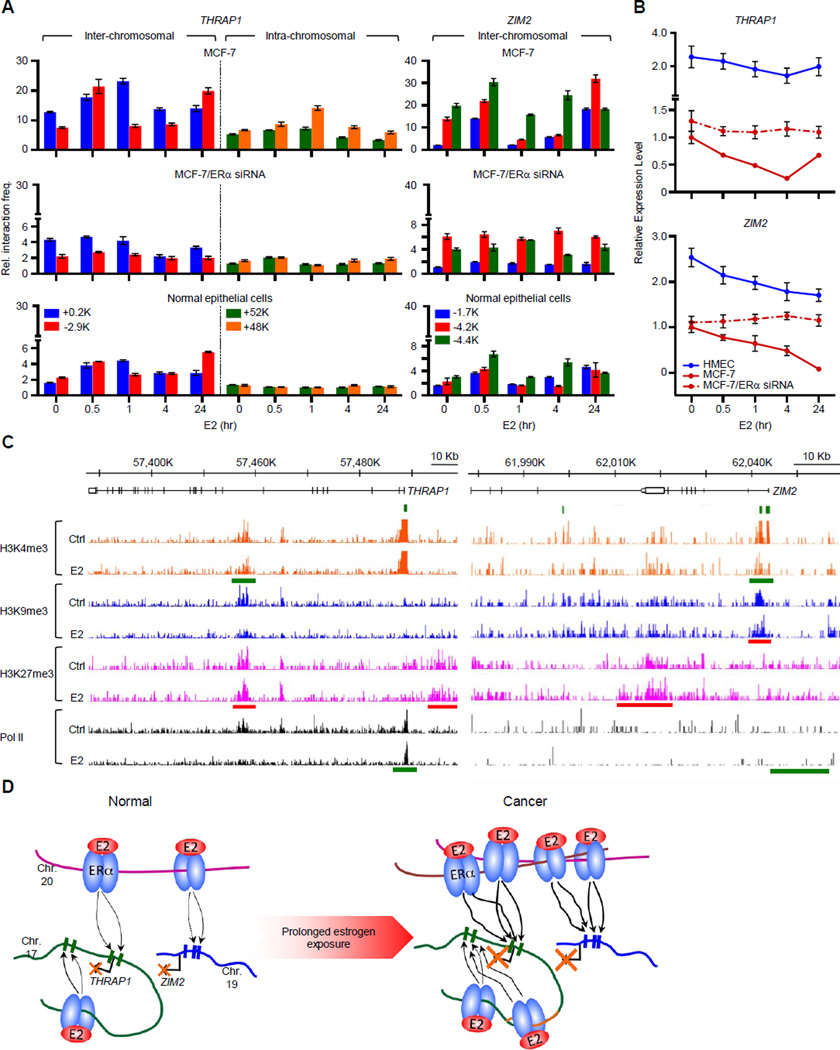
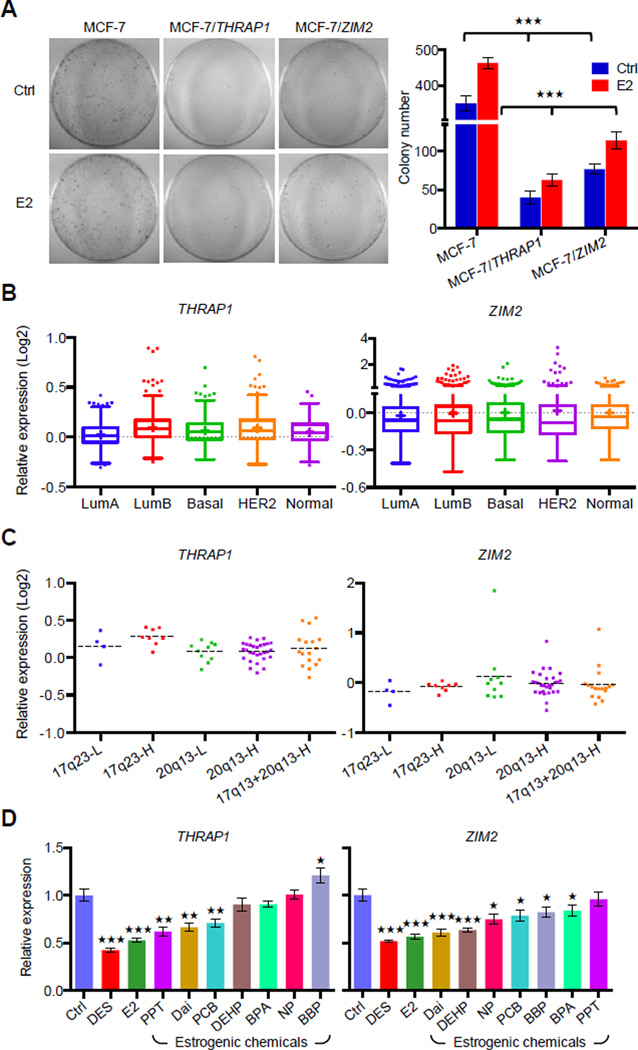
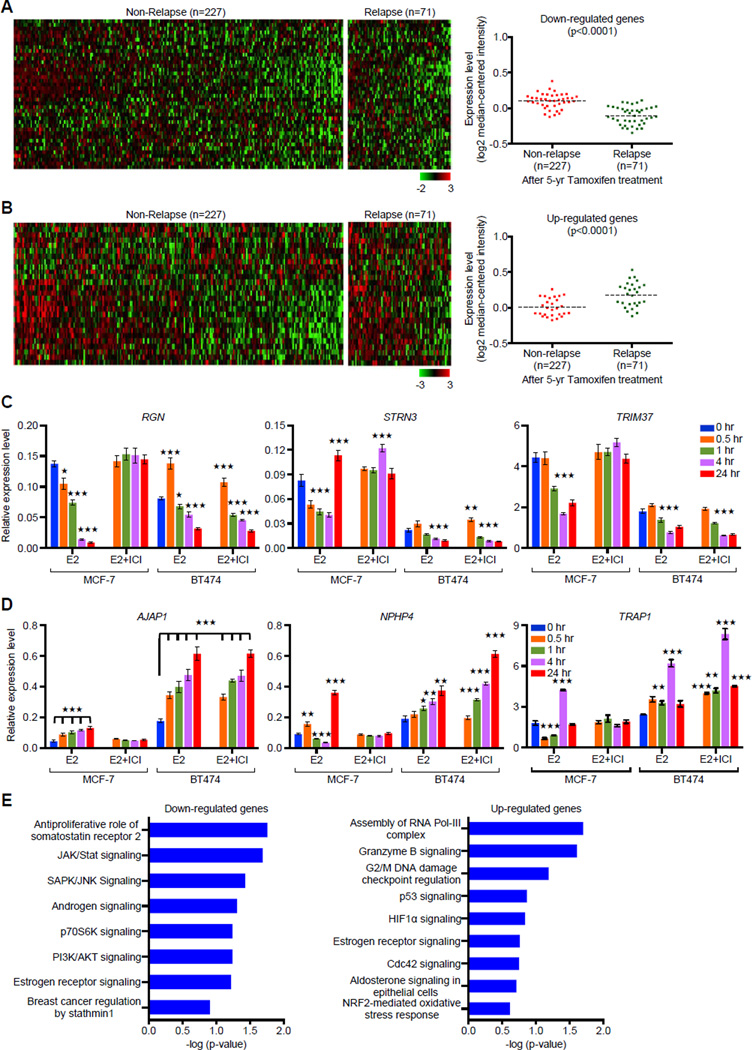
A causal role of gene amplification in tumorigenesis is well known, whereas amplification of DNA regulatory elements as an oncogenic driver remains unclear. In this study, we integrated next-generation sequencing approaches to map distant estrogen response elements (DEREs) that remotely control the transcription of target genes through chromatin proximity. Two densely mapped DERE regions located on chromosomes 17q23 and 20q13 were frequently amplified in estrogen receptor-α-positive luminal breast cancer. These aberrantly amplified DEREs deregulated target gene expression potentially linked to cancer development and tamoxifen resistance. Progressive accumulation of DERE copies was observed in normal breast progenitor cells chronically exposed to estrogenic chemicals. These findings may extend to other DNA regulatory elements, the amplification of which can profoundly alter target transcriptome during tumorigenesis.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
References
-
- Albertson DG. Gene amplification in cancer. Trends Genet. 2006;22:447–455. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
