

Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin
- PMID: 23954158
- PMCID: PMC3770529
- DOI: 10.1016/j.chom.2013.07.012
Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin
Abstract
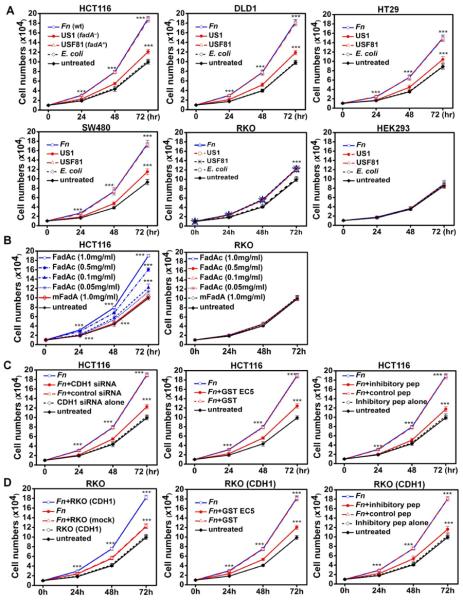
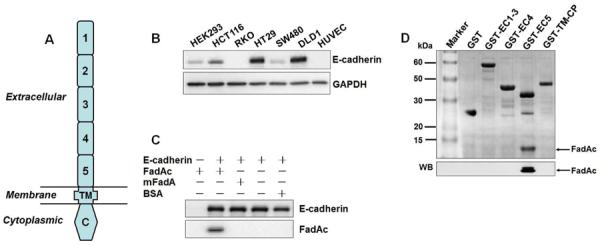
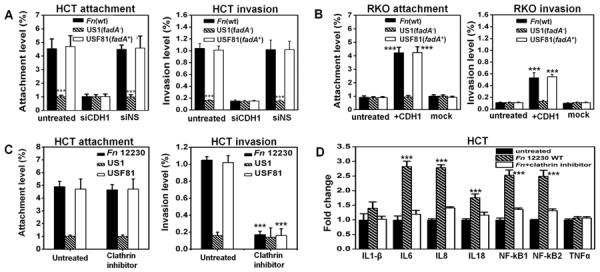
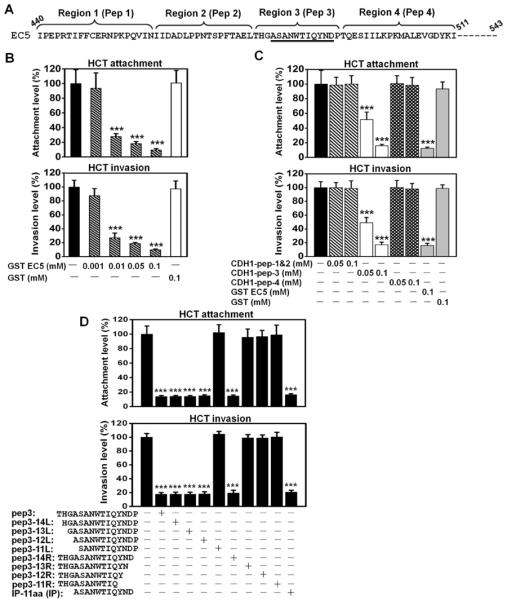
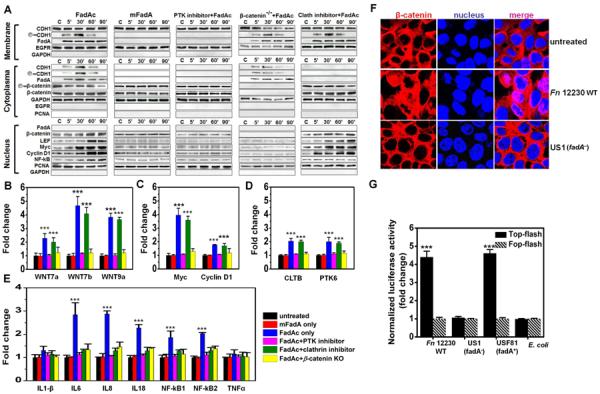
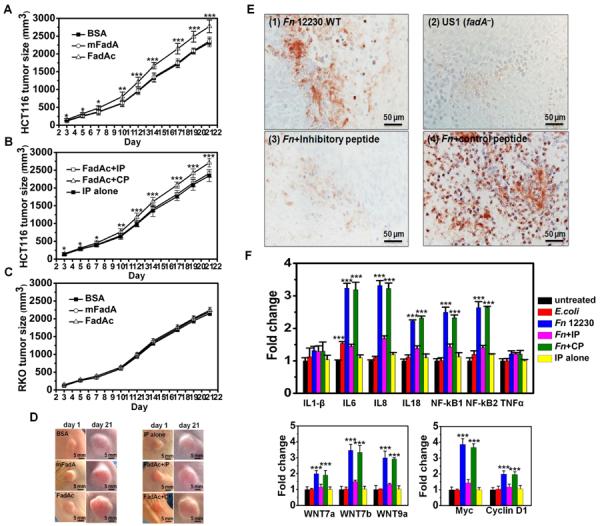
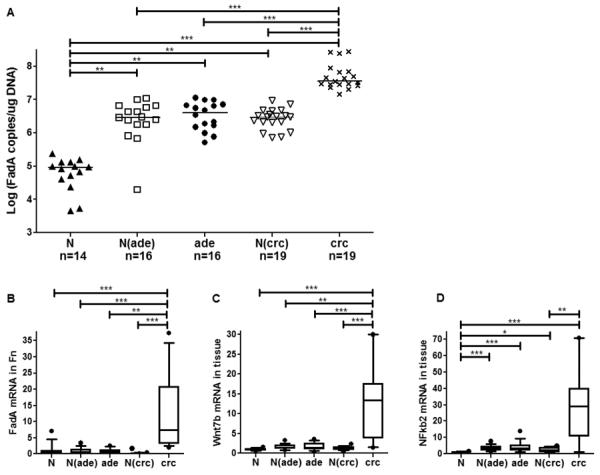
Fusobacterium nucleatum (Fn) has been associated with colorectal cancer (CRC), but causality and underlying mechanisms remain to be established. We demonstrate that Fn adheres to, invades, and induces oncogenic and inflammatory responses to stimulate growth of CRC cells through its unique FadA adhesin. FadA binds to E-cadherin, activates β-catenin signaling, and differentially regulates the inflammatory and oncogenic responses. The FadA-binding site on E-cadherin is mapped to an 11-amino-acid region. A synthetic peptide derived from this region of E-cadherin abolishes FadA-induced CRC cell growth and oncogenic and inflammatory responses. The fadA gene levels in the colon tissue from patients with adenomas and adenocarcinomas are >10-100 times higher compared to normal individuals. The increased FadA expression in CRC correlates with increased expression of oncogenic and inflammatory genes. This study unveils a mechanism by which Fn can drive CRC and identifies FadA as a potential diagnostic and therapeutic target for CRC.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Fusobacterium spp. and colorectal cancer: cause or consequence?Trends Microbiol. 2013 Oct;21(10):506-8. doi: 10.1016/j.tim.2013.08.004. Epub 2013 Sep 9. Trends Microbiol. 2013. PMID: 24029382 Free PMC article.
References
-
- ACS Cancer Facts & Figures 2012. American Cancer Society (ACS) 2012:1–66.
-
- Baron JA, Sandler RS. Nonsteroidal anti-inflammatory drugs and cancer prevention. Annual review of medicine. 2000;51:511–523. - PubMed
-
- Bartlett DL, Chu E. Can metastatic colorectal cancer be cured? Oncology. 2012;26:266–275. - PubMed
-
- Bryant DM, Stow JL. The ins and outs of E-cadherin trafficking. Trends in cell biology. 2004;14:427–434. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous