

Review
doi: 10.1007/s00109-013-1074-5.
Epub 2013 Aug 17.
Pathologies at the nexus of blood coagulation and inflammation: thrombin in hemostasis, cancer, and beyond
Affiliations
- PMID: 23955016
- PMCID: PMC3825489
- DOI: 10.1007/s00109-013-1074-5
Item in Clipboard
Review
Pathologies at the nexus of blood coagulation and inflammation: thrombin in hemostasis, cancer, and beyond
J Mol Med (Berl).
2013 Nov.
Abstract
Thrombin is the protease involved in blood coagulation. Its deregulation can lead to hemostatic abnormalities, which range from subtle subclinical to serious life-threatening coagulopathies, i.e., during septicemia. Additionally, thrombin plays important roles in many (patho)physiological conditions that reach far beyond its well-established role in stemming blood loss and thrombosis, including embryonic development and angiogenesis but also extending to inflammatory processes, complement activation, and even tumor biology. In this review, we will address thrombin's broad roles in diverse (patho)physiological processes in an integrative way. We will also discuss thrombin as an emerging major target for novel therapies.
Figures
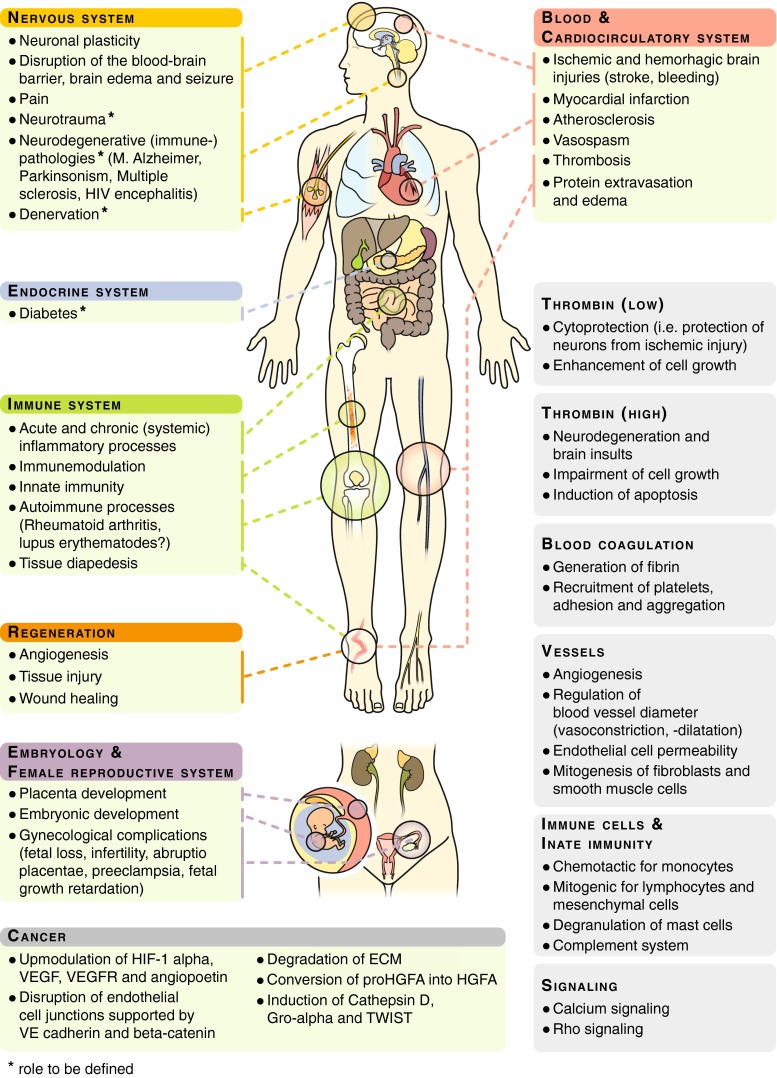
Thrombin and thrombin functions in development, physiology, and pathophysiology
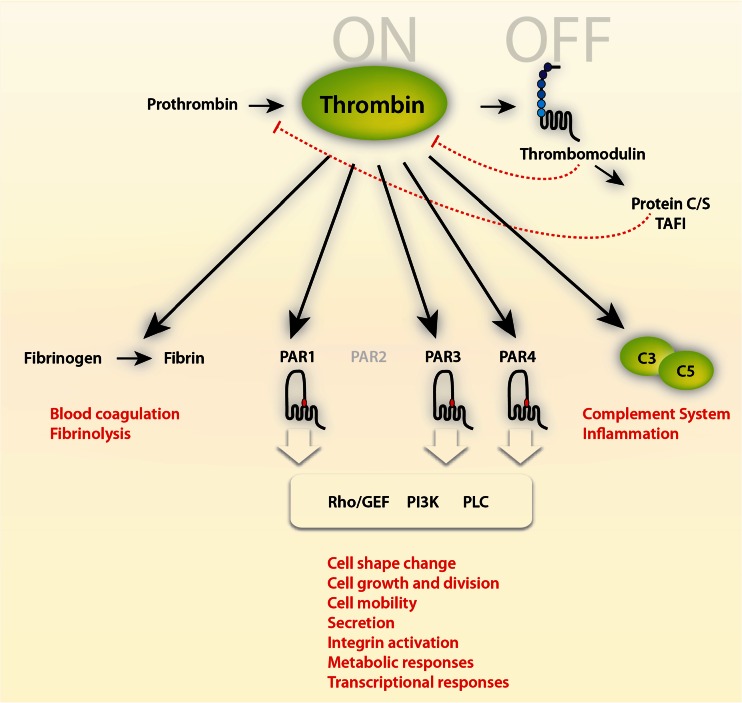
Mechanisms of thrombin action. Thrombin is a multifunctional serine protease involved in blood coagulation, complement activation, and numerous cellular functions mediated via G protein-coupled prote'ase-activated receptor (PAR) signaling pathways (for further details, see [11]). Thrombin is antagonized by binding to thrombomodulin, a multi-domain proteoglycan found primarily on endothelial cells (see natural inhibitors of thrombin, Table 1). However, thrombin bound to thrombomodulin augments the ability to activate protein C, a natural anticoagulant, which in a negative feedback loop represses the generation of thrombin (protein C itself also has antiapoptotic and anti-inflammatory activity and increases activation of thrombin-activatable fibrinolysis inhibitor (TAFI), an enzyme which blocks the activation of plasminogen and inactivates vasoactive peptides like complement C5a, not shown)
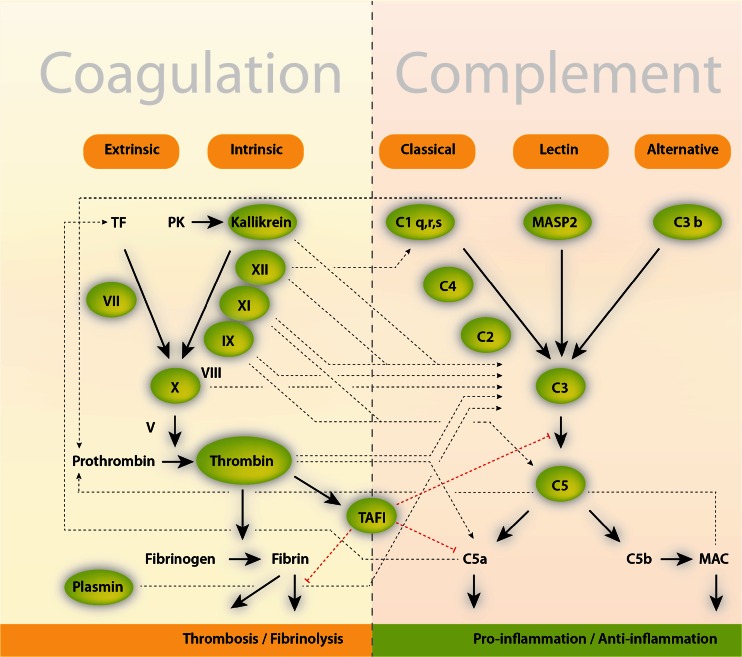
Cross talk between the coagulation and complement system. The coagulation cascade, the complement system, and fibrinolysis (simplified) communicate through many direct and bidirectional interactions (indicated). Activated clotting Factor XII can activate the classical complement pathway through cleavage of the complement component C1. Similarly, thrombin, kallikrein, and plasmin directly cleave complement component C3, as well as its activation fragments (not shown). Moreover, thrombin can cleave C5 into C5a, which occurs independently of C3 and therefore represents a bypass of the three traditional complement activation pathways (the classical, the lectin, and alternative pathways) [3]. Thrombin-activatable fibrinolysis inhibitor (TAFI) inactivates C3a and C5a in a negative feedback loop. The complement system also amplifies coagulation through the C5a-mediated induction of expression of tissue factor (TF) and plasminogen activator inhibitor 1 by leukocytes (not shown), the latter of which inhibits fibrinolysis. In addition, mannan-binding lectin serine protease 2 (MASP2) of the lectin complement activation pathway triggers coagulation by converting prothrombin to thrombin. MAC, membrane attack complex (C5b–C9); see also [3, 161]
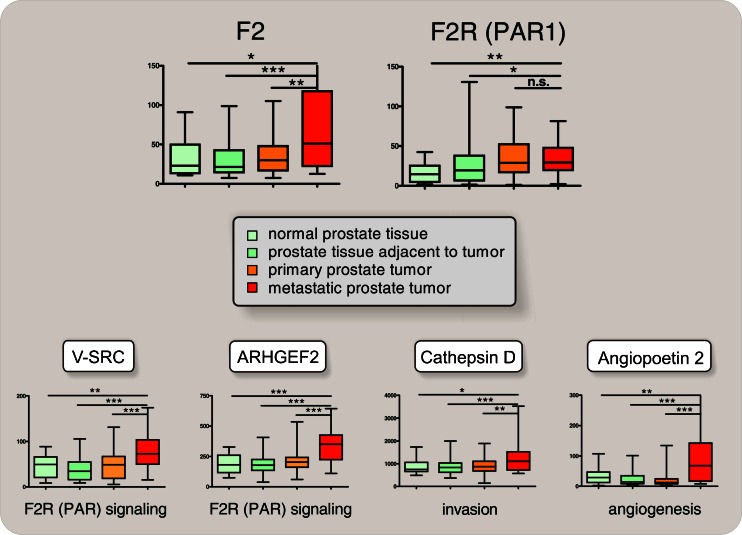
Stage-dependent induction of ectopic (i.e., extrahepatic) thrombin (F2) gene expression in metastatic prostate cancer. Normalized mRNA expression of the thrombin (F2) gene, of the F2 Receptor (F2R/PAR1), of V-SRC and ARHGEF2 (surrogate for activated F2R signaling), and of cathepsin D and angiopoetin 2 (for invasion and angiogenesis [6, 137]), obtained from gene expression profiling after extraction, normalization, and reassembly of 171 human samples (see [162] GEO GDS2545 record) including metastatic prostate cancer tissues (n = 24; GSE6605), nonmetastatic primary prostate tumors (n = 60; GSE6606), prostate tissues adjacent to the tumor (n = 63; GSE6608), and normal donor prostate tissues (n = 18, GSE6604) [163, 164] (median, horizontal line; 25th through 75th percentile, box; range, standard error of the mean (SEM); *p < 0.05; **p < 0.01; ***p < 0.001)
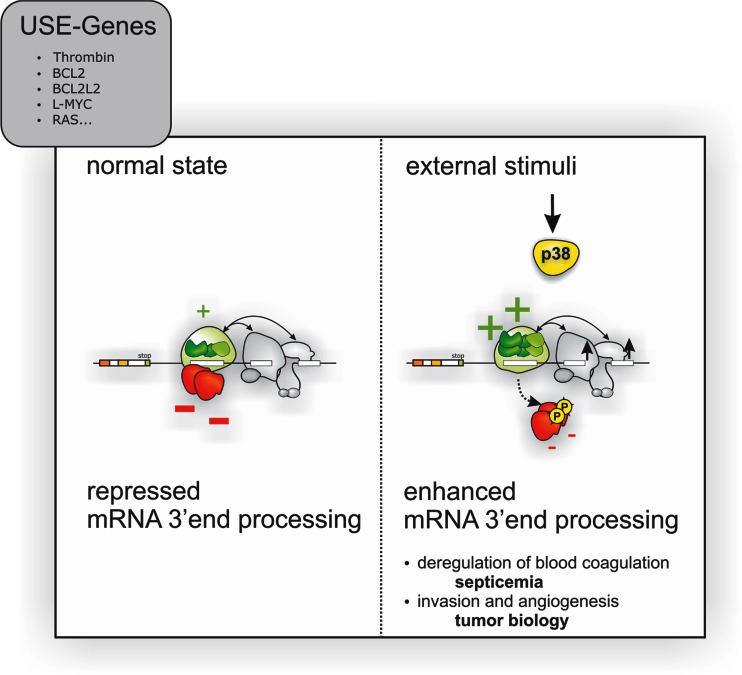
Extracellular stimuli induce thrombin gene expression by p38 MAPK activation. Extracellular stimuli activate p38 MAPK and thereby phosphorylate regulatory proteins (red), which “catalyzes” the remodeling of a stimulatory ribonucleoprotein (RNP) complexes (green) to up-modulate the efficiency of thrombin mRNA 3′ end processing. This mechanism allows modulating cellular functions, such as blood coagulation by controlling the amount of thrombin protein produced. Yet activation of this mechanism also appears to play an important role in other pathophysiological processes (such as tumorigenesis) and drives cellular programs involved in tumor invasion and neoangiogenesis by the activation of thrombin receptor signaling (F2R, PAR-1) and degradation of extracellular matrix (figure adopted from Cell Press [113])
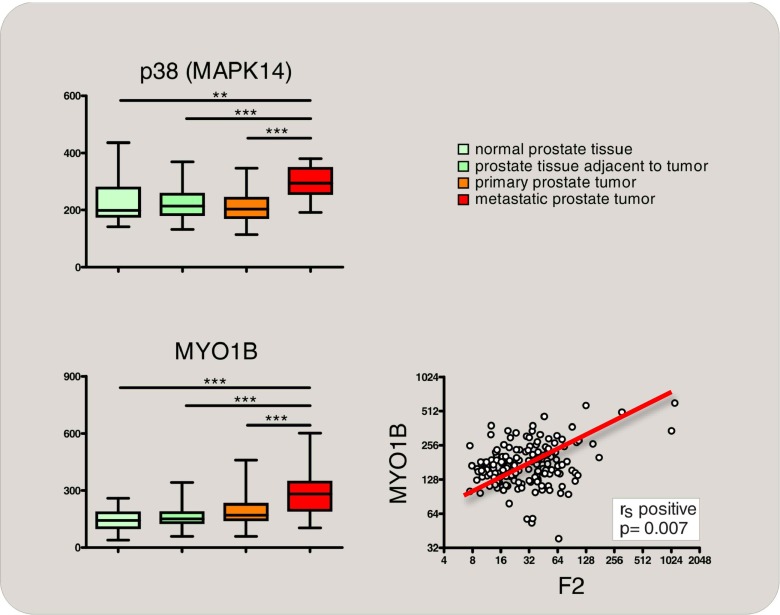
Induction of thrombin (F2) gene expression correlates with a stage-dependent activation of p38 MAPK signaling in metastatic prostate cancer. Normalized mRNA expression of p38 MAPK (upper diagram) and of MYO1B (lower left diagram), which reflects activation of p38 MAPK signaling [165], obtained from gene expression profiling of 171 human samples [162] (median, horizontal line; 25th through 75th percentile, box; range, standard error of the mean (SEM); *p < 0.05; **p < 0.01; ***p < 0.001). Correlation of p38 MAPK signaling activation (MYO1B gene expression) with F2 gene expression is shown in the lower right diagram (Spearman’s rank correlation)
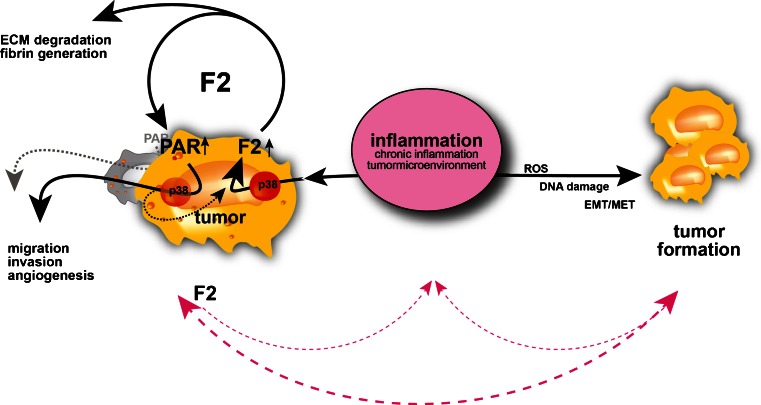
Model for inflammation as a unifying trigger predisposing to deregulated blood coagulation (thrombin gene expression) and tumor formation. Inflammatory stimuli can induce both tumor formation (simplified) and thrombin (F2) gene expression. This in turn leads to a disequilibrium of pro- and anticoagulatory activities (and thereby promotes tumor-associated thrombus formation) and drives protumorigenic cellular programs (in an autocrine and/or paracrine manner; SD unpublished). Tumor formation will thus be supported by the tumor-promoting properties of thrombin; vice versa, tumor formation elicits detrimental inflammatory responses [159], which in turn further promote tumorigenesis and p38 MAPK (p38)-mediated induction of thrombin gene expression. (Extracellular matrix (ECM), Reactive oxygen species (ROS), epithelial–mesenchymal transformation, mesenchymal–epithelial transformation (EMT/MET))
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
