

Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita
- PMID: 23956869
- PMCID: PMC3727188
- DOI: 10.1155/2013/812029
Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita
Abstract
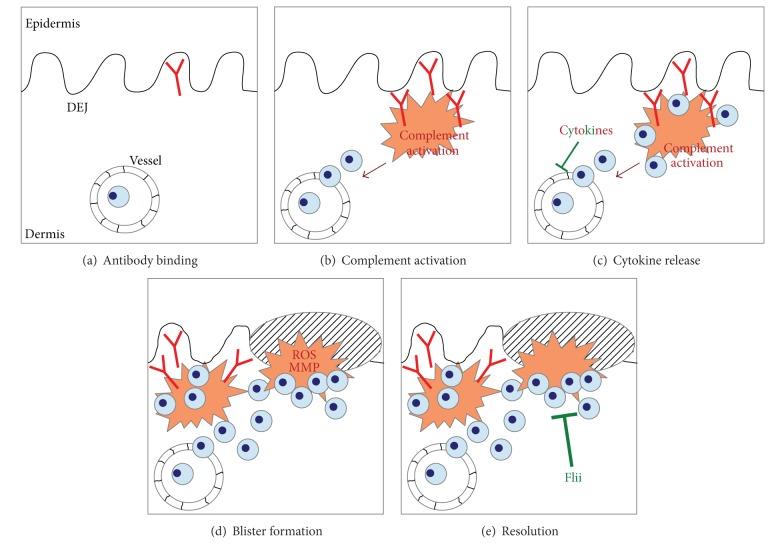
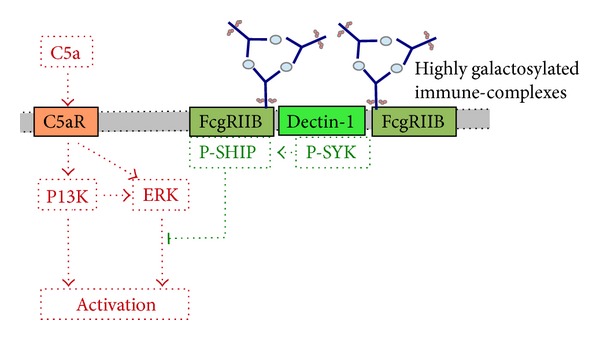
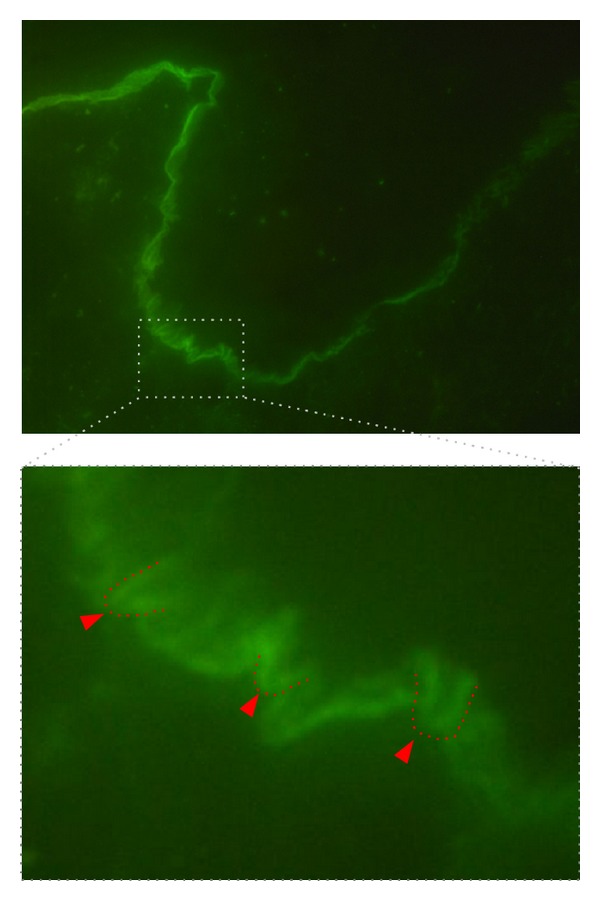
Epidermolysis bullosa acquisita (EBA) is a chronic mucocutaneous autoimmune skin blistering disease. The pathogenic relevance of autoantibodies targeting type VII collagen (COL7) has been well-documented. Therefore, EBA is a prototypical autoimmune disease with a well-characterized pathogenic relevance of autoantibody binding to the target antigen. EBA is a rare disease with an incidence of 0.2 new cases per million and per year. The current treatment of EBA relies on general immunosuppressive therapy, which does not lead to remission in all cases. Therefore, there is a high, so far unmet medical need for the development of novel therapeutic options. During the last 10 years, several novel in vitro and in vivo models of EBA have been established. These models demonstrated a critical role of the genetic background, T cells, and cytokines for mediating the loss of tolerance towards COL7. Neutrophils, complement activation, Fc gamma receptor engagement, cytokines, several molecules involved in cell signaling, release of reactive oxygen species, and matrix metalloproteinases are crucial for autoantibody-induced tissue injury in EBA. Based on this growing understanding of the diseases' pathogenesis, several potential novel therapeutic targets have emerged. In this review, the clinical presentation, pathogenesis, diagnosis, and current treatment options for EBA are discussed in detail.
Figures
Similar articles
-
Model systems duplicating epidermolysis bullosa acquisita: a methodological review.Autoimmunity. 2012 Feb;45(1):102-10. doi: 10.3109/08916934.2011.606451. Epub 2011 Sep 19. Autoimmunity. 2012. PMID: 21923614 Review.
-
Inhibition of interferon gamma impairs induction of experimental epidermolysis bullosa acquisita.Front Immunol. 2024 May 10;15:1343299. doi: 10.3389/fimmu.2024.1343299. eCollection 2024. Front Immunol. 2024. PMID: 38799441 Free PMC article.
-
Fcγ Receptor IIB Controls Skin Inflammation in an Active Model of Epidermolysis Bullosa Acquisita.Front Immunol. 2020 Jan 14;10:3012. doi: 10.3389/fimmu.2019.03012. eCollection 2019. Front Immunol. 2020. PMID: 31993051 Free PMC article.
-
Heat shock protein 90 is required for ex vivo neutrophil-driven autoantibody-induced tissue damage in experimental epidermolysis bullosa acquisita.Exp Dermatol. 2015 Jun;24(6):471-3. doi: 10.1111/exd.12680. Epub 2015 Mar 25. Exp Dermatol. 2015. PMID: 25739426
-
Immune mechanism-targeted treatment of experimental epidermolysis bullosa acquisita.Expert Rev Clin Immunol. 2015;11(12):1365-78. doi: 10.1586/1744666X.2015.1085801. Epub 2015 Oct 15. Expert Rev Clin Immunol. 2015. PMID: 26471717 Review.
Cited by
-
Structural and biophysical characterization of the type VII collagen vWFA2 subdomain leads to identification of two binding sites.FEBS Open Bio. 2020 Apr;10(4):580-592. doi: 10.1002/2211-5463.12807. Epub 2020 Mar 14. FEBS Open Bio. 2020. PMID: 32031736 Free PMC article.
-
Reduced skin blistering in experimental epidermolysis bullosa acquisita after anti-TNF treatment.Mol Med. 2017 Feb;22:918-926. doi: 10.2119/molmed.2015.00206. Epub 2016 Dec 20. Mol Med. 2017. PMID: 27999842 Free PMC article.
-
Diffuse Bullous Eruptions in an Elderly Woman: Late-Onset Bullous Systemic Lupus Erythematosus.Case Rep Dermatol. 2016 Oct 13;8(3):278-282. doi: 10.1159/000448392. eCollection 2016 Sep-Dec. Case Rep Dermatol. 2016. PMID: 27920678 Free PMC article.
-
Cytoskeletal Regulation of Inflammation and Its Impact on Skin Blistering Disease Epidermolysis Bullosa Acquisita.Int J Mol Sci. 2016 Jul 13;17(7):1116. doi: 10.3390/ijms17071116. Int J Mol Sci. 2016. PMID: 27420054 Free PMC article. Review.
-
Role of immune cells in the ocular manifestations of pemphigoid diseases.Ther Adv Ophthalmol. 2019 Aug 8;11:2515841419868128. doi: 10.1177/2515841419868128. eCollection 2019 Jan-Dec. Ther Adv Ophthalmol. 2019. PMID: 31448360 Free PMC article. Review.
References
-
- Elliott GT. Two cases of epidermolysis bullosa. Journal of Cutaneous and Genito-Urinary Diseases. 1895;13(article 10)
-
- Roenigk HH, Jr., Ryan JG, Bergfeld WF. Epidermolysis bullosa acquisita. Report of three cases and review of all published cases. Archives of Dermatology. 1971;103(1):1–10. - PubMed
-
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? Journal of Dermatology. 2010;37(3):220–230. - PubMed
-
- Schmidt E, Zillikens D. Pemphigoid diseases. The Lancet. 2013;381:320–332. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources