

Impact of the gut microbiome on mucosal inflammation
- PMID: 23957963
- PMCID: PMC3773492
- DOI: 10.1016/j.it.2013.07.001
Impact of the gut microbiome on mucosal inflammation
Abstract
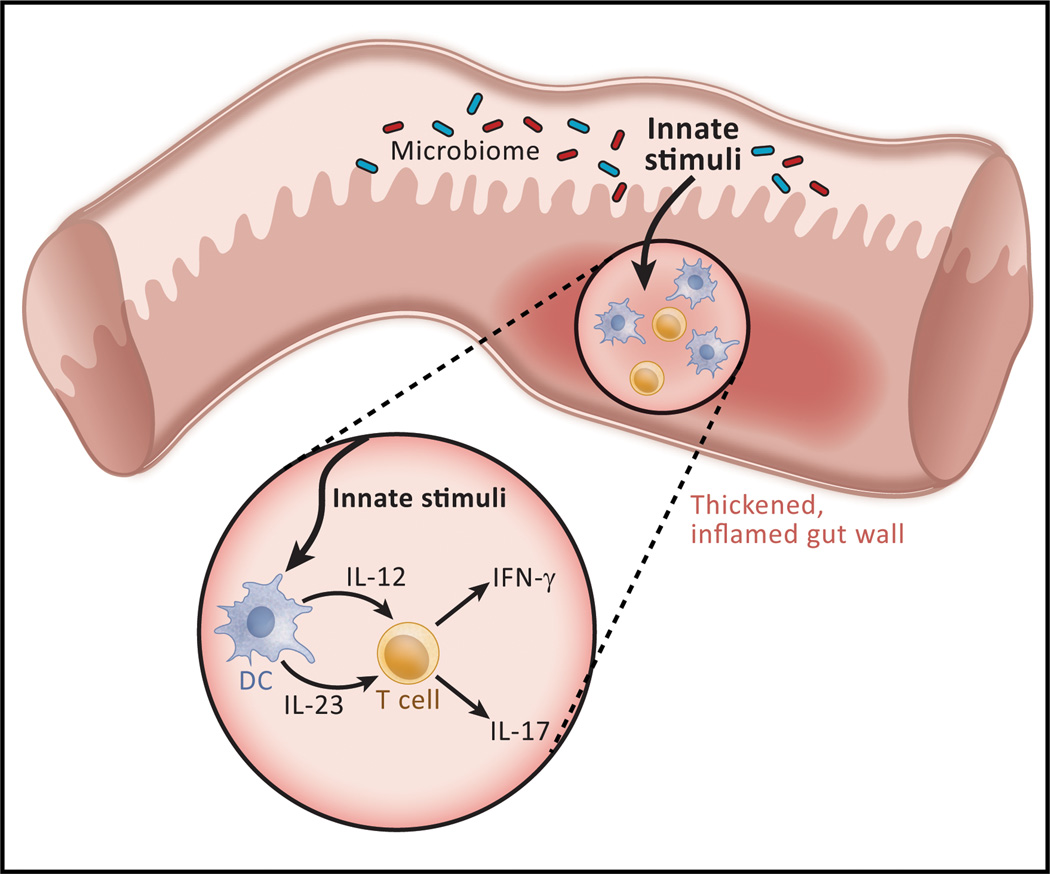
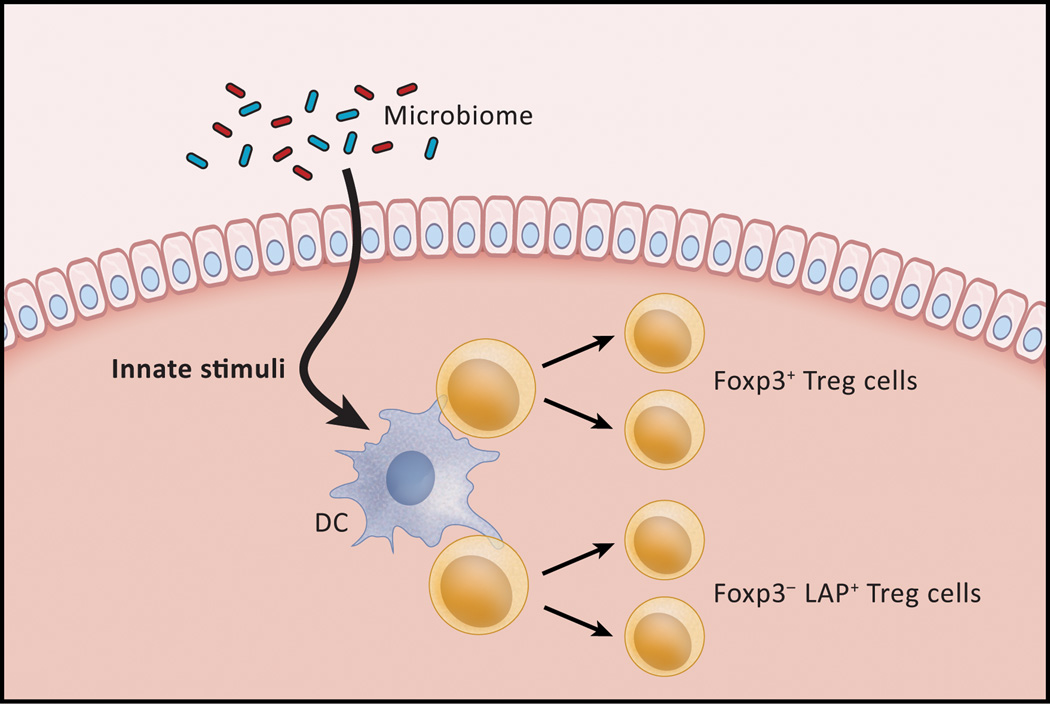
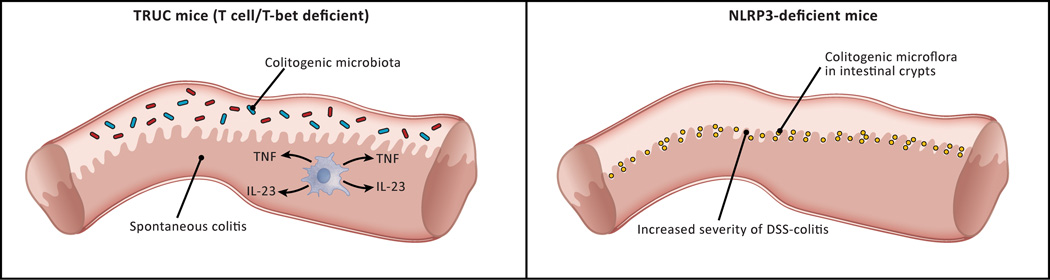
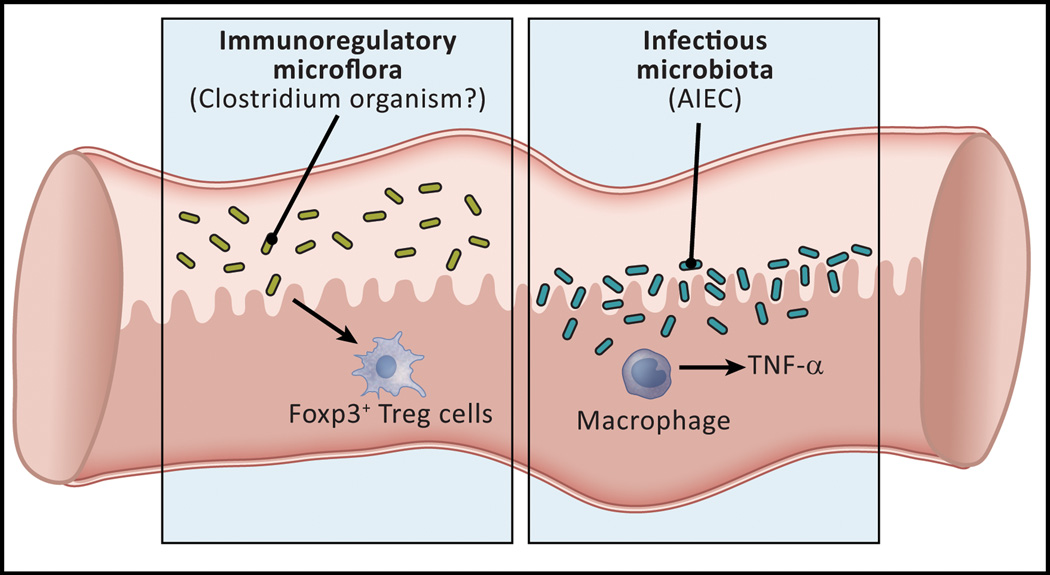
In the past 10 years it has become increasingly apparent that the gut microbiome has profound effects on the immune system to which it is juxtaposed, the mucosal immune system. Here, I explore recent studies in which the effects of the microbiota expand or facilitate anti-inflammatory or regulatory immunological machinery or which favor development of proinflammatory immunological machinery in this system. I then focus on how these opposing processes play out in inflammatory bowel disease (IBD); a disease in which normal immune homeostasis is disturbed and inflammation takes hold.
Keywords: Anti-inflammatory organisms; Gut microbiome; Inflammatory Bowel Disease; Mucosal regulatory T cells; Proinflammatory (colitogenic organisms).
Published by Elsevier Ltd.
Figures
References
-
- Strober W, Fuss IJ, Blumberg RS. The immunology of mucosal models of inflammation. Annu. Rev. Immunol. 2001;20:495–549. - PubMed
-
- Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujmura T, Takeuchi O, Yoshimori T, Akira S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
