

Glycan-based biomarkers for mucopolysaccharidoses
- PMID: 23958290
- PMCID: PMC3769472
- DOI: 10.1016/j.ymgme.2013.07.016
Glycan-based biomarkers for mucopolysaccharidoses
Abstract
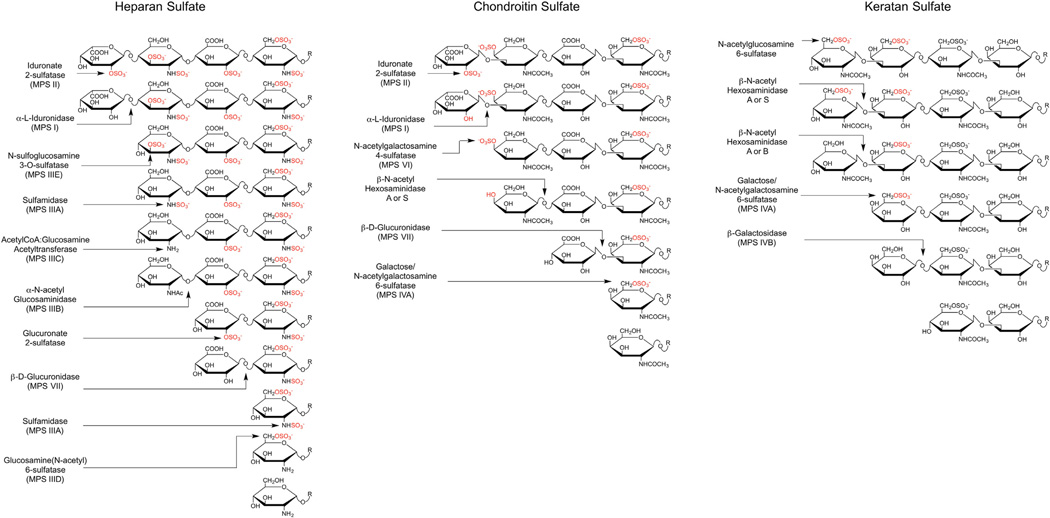
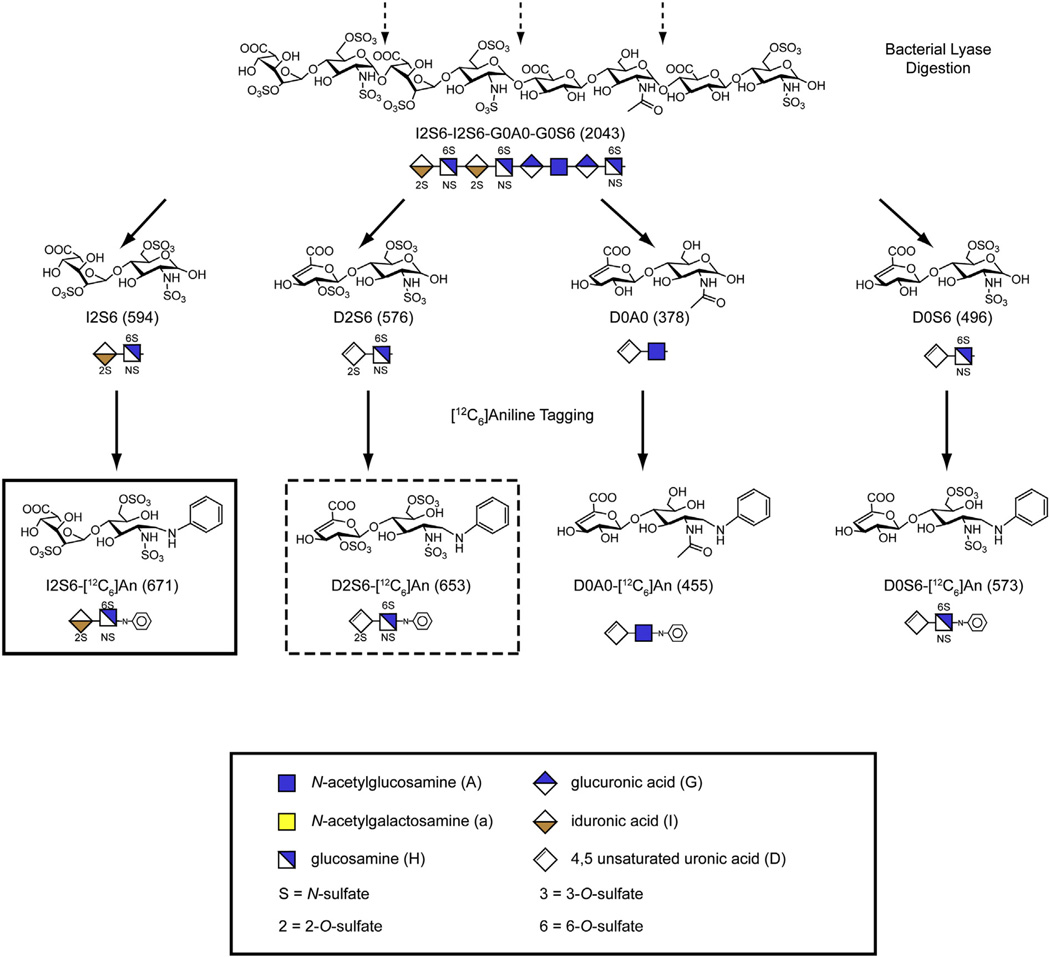
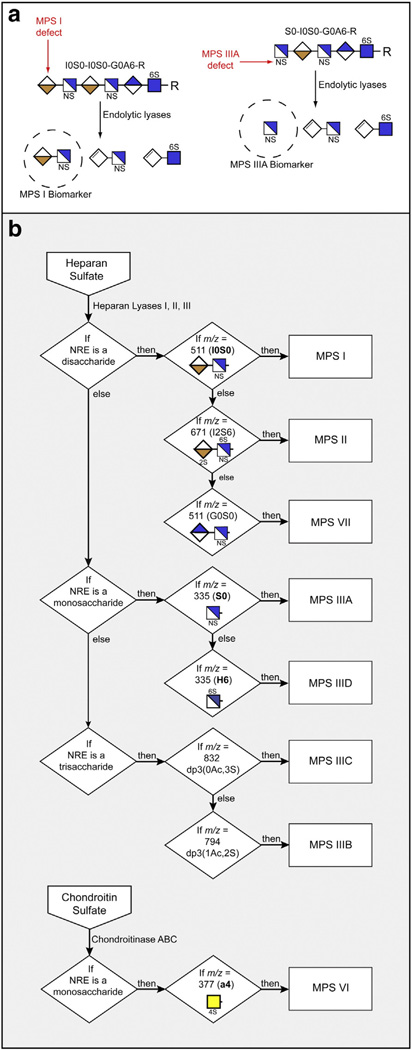
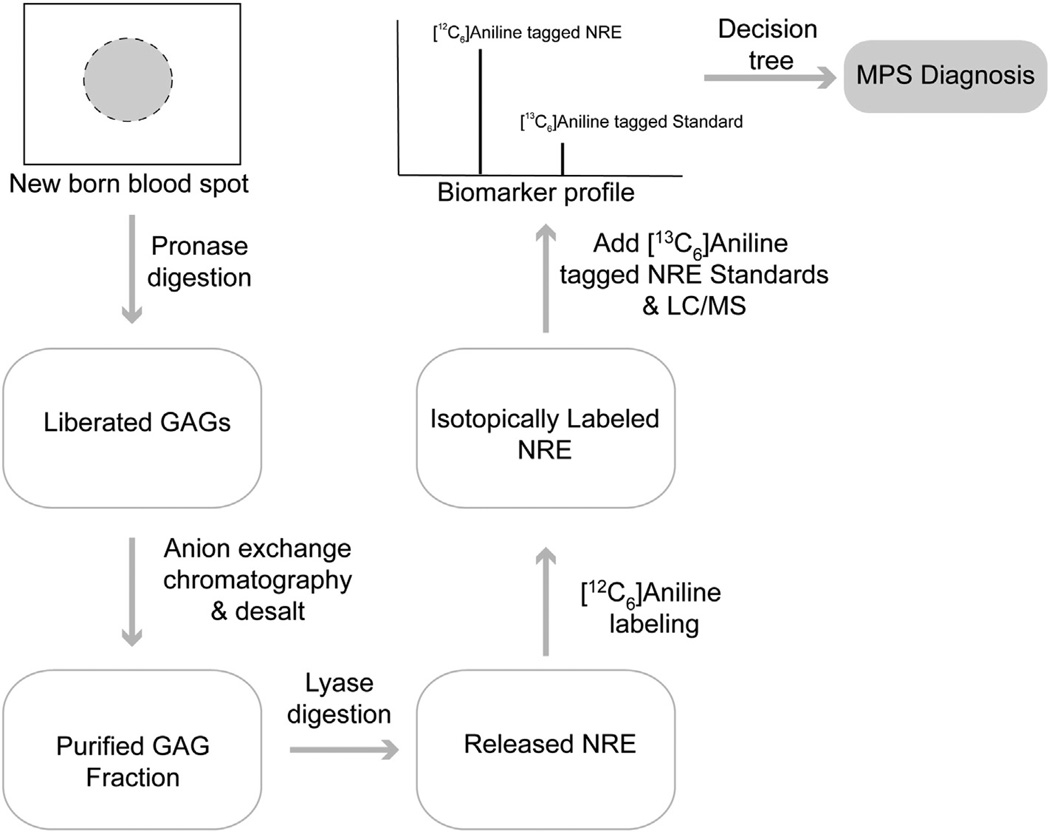
The mucopolysaccharidoses (MPS) result from attenuation or loss of enzyme activities required for lysosomal degradation of the glycosaminoglycans, hyaluronan, heparan sulfate, chondroitin/dermatan sulfate, and keratan sulfate. This review provides a summary of glycan biomarkers that have been used to characterize animal models of MPS, for diagnosis of patients, and for monitoring therapy based on hematopoietic stem cell transplantation and enzyme replacement therapy. Recent advances have focused on the non-reducing terminus of the glycosaminoglycans that accumulate as biomarkers, using a combination of enzymatic digestion with bacterial enzymes followed by quantitative liquid chromatography/mass spectrometry. These new methods provide a simple, rapid diagnostic strategy that can be applied to samples of urine, blood, cerebrospinal fluid, cultured cells and dried blood spots from newborn infants. Analysis of the non-reducing end glycans provides a method for monitoring enzyme replacement and substrate reduction therapies and serves as a discovery tool for uncovering novel biomarkers and new forms of mucopolysaccharidoses.
Keywords: Carbohydrate biomarkers; Glycosaminoglycans; Lysosomal storage disorders; Mass spectrometry; Mucopolysaccharidoses; Sensi-Pro assay.
© 2013 Elsevier Inc. All rights reserved.
Conflict of interest statement
Jillian R. Brown and Brett E. Crawford were employees of Zacharon Pharmaceuticals, Inc. at the time that the paper was written and Roger Lawrence and Jeffrey D. Esko were paid consultants to the company.
Figures
References
-
- Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. Lysosomal Disorders. Metabolic and Molecular Basis of Inherited Disease. San Francisco: MacGraw-Hill; 2001. pp. 3371–3896.
-
- Bouwman MG, Teunissen QG, FA Wijburg, Linthorst GE. ‘Doctor Google’ ending the diagnostic odyssey in lysosomal storage disorders: parents using internet search engines as an efficient diagnostic strategy in rare diseases. Arch. Dis. Child. 2010;95:642–644. - PubMed
-
- Neufeld EF, Muenzer J. The Mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. Metabolic and Molecular Basis of Inherited Disease. San Francisco: MacGraw-Hill; 2001. pp. 3421–3452.
-
- Kaneiwa T, Mizumoto S, Sugahara K, Yamada S. Identification of human hyaluronidase-4 as a novel chondroitin sulfate hydrolase that preferentially cleaves the galactosaminidic linkage in the trisulfated tetrasaccharide sequence. Glycobiology. 2010;20:300–309. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
