

Evading apoptosis in cancer
- PMID: 23958396
- PMCID: PMC4091735
- DOI: 10.1016/j.tcb.2013.07.006
Evading apoptosis in cancer
Abstract
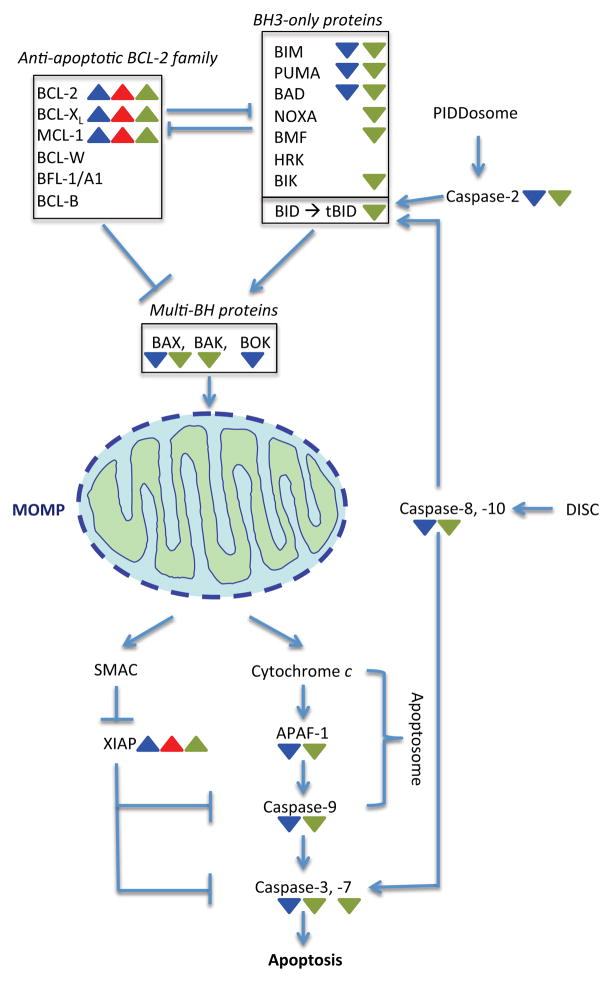
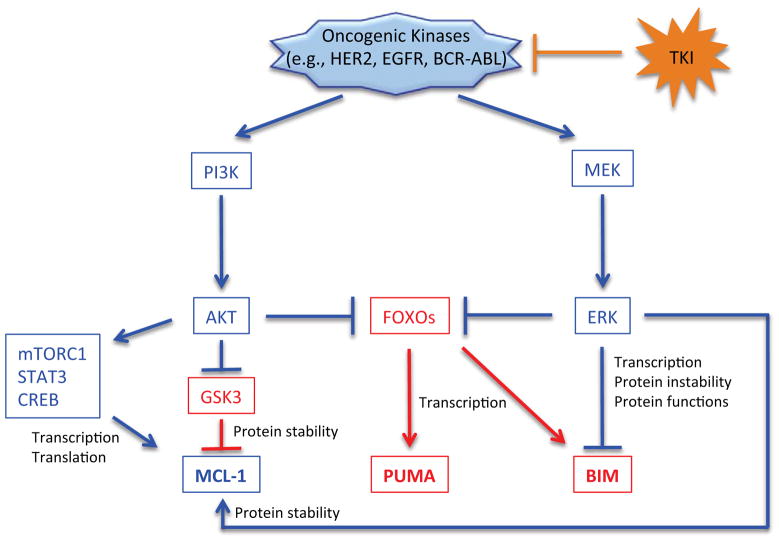
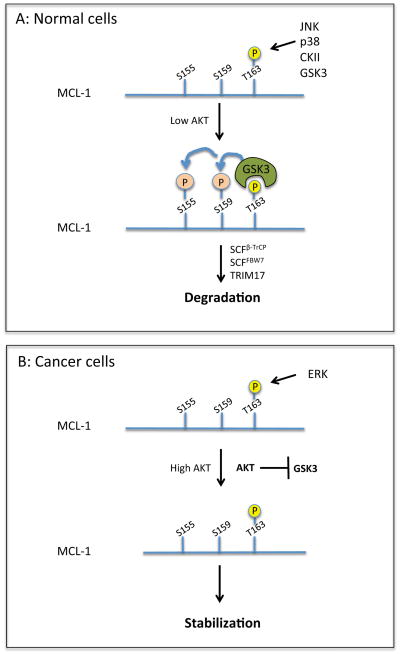
Carcinogenesis is a mechanistically complex and variable process with a plethora of underlying genetic causes. Cancer development comprises a multitude of steps that occur progressively starting with initial driver mutations leading to tumorigenesis and, ultimately, metastasis. During these transitions, cancer cells accumulate a series of genetic alterations that confer on the cells an unwarranted survival and proliferative advantage. During the course of development, however, cancer cells also encounter a physiologically ubiquitous cellular program that aims to eliminate damaged or abnormal cells: apoptosis. Thus, it is essential that cancer cells acquire instruments to circumvent programmed cell death. Here we discuss emerging evidence indicating how cancer cells adopt various strategies to override apoptosis, including amplifying the antiapoptotic machinery, downregulating the proapoptotic program, or both.
Keywords: BH3; MOMP; caspase; phosphorylation; ubiquitination.
Copyright © 2013 Elsevier Ltd. All rights reserved.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
