

Considerations for normalization of DNA methylation data by Illumina 450K BeadChip assay in population studies
- PMID: 23959097
- PMCID: PMC6242262
- DOI: 10.4161/epi.26037
Considerations for normalization of DNA methylation data by Illumina 450K BeadChip assay in population studies
Abstract
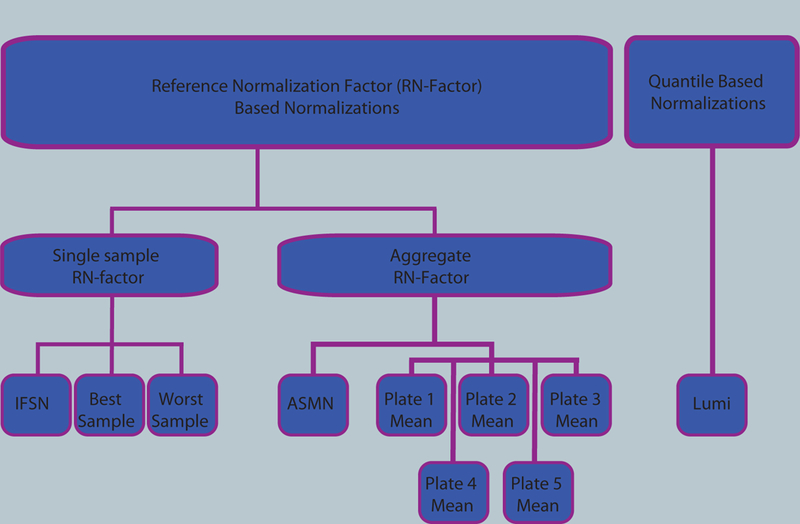
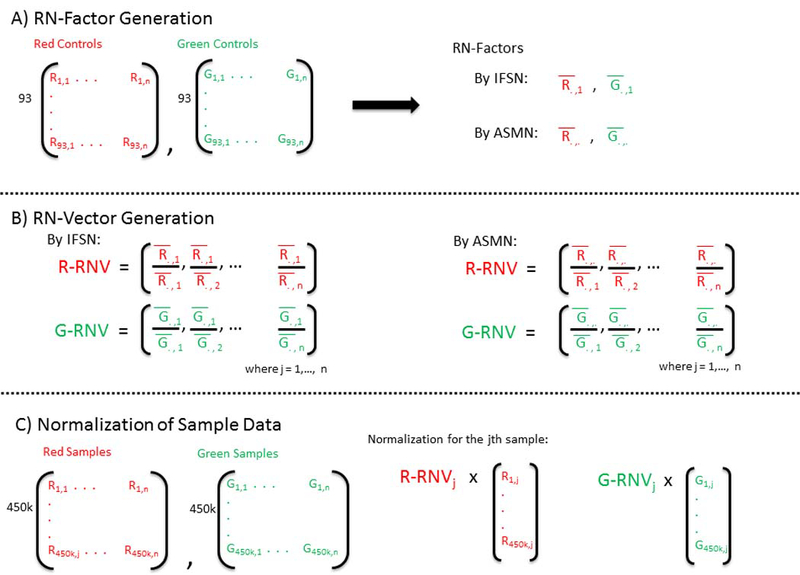
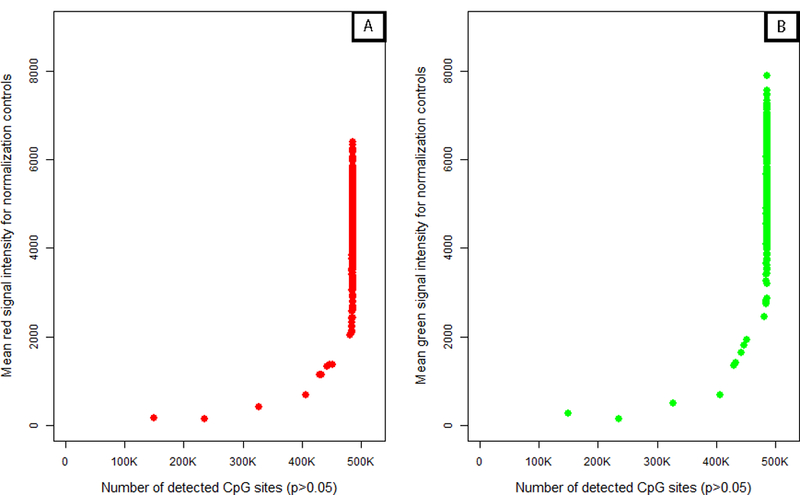
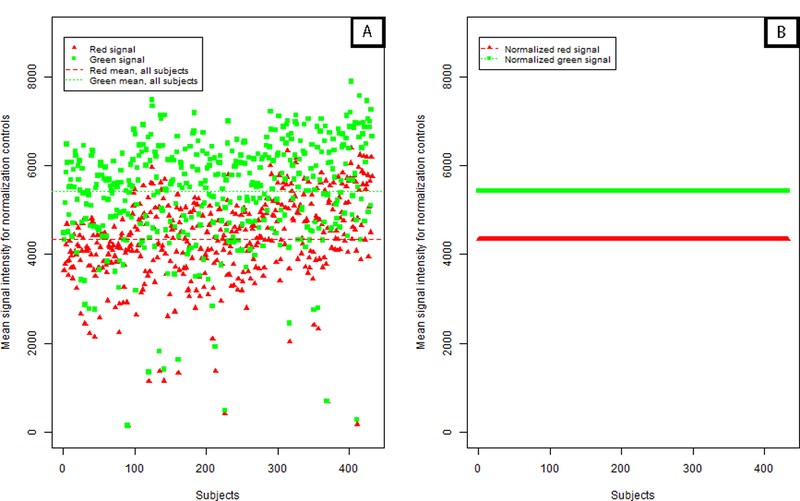
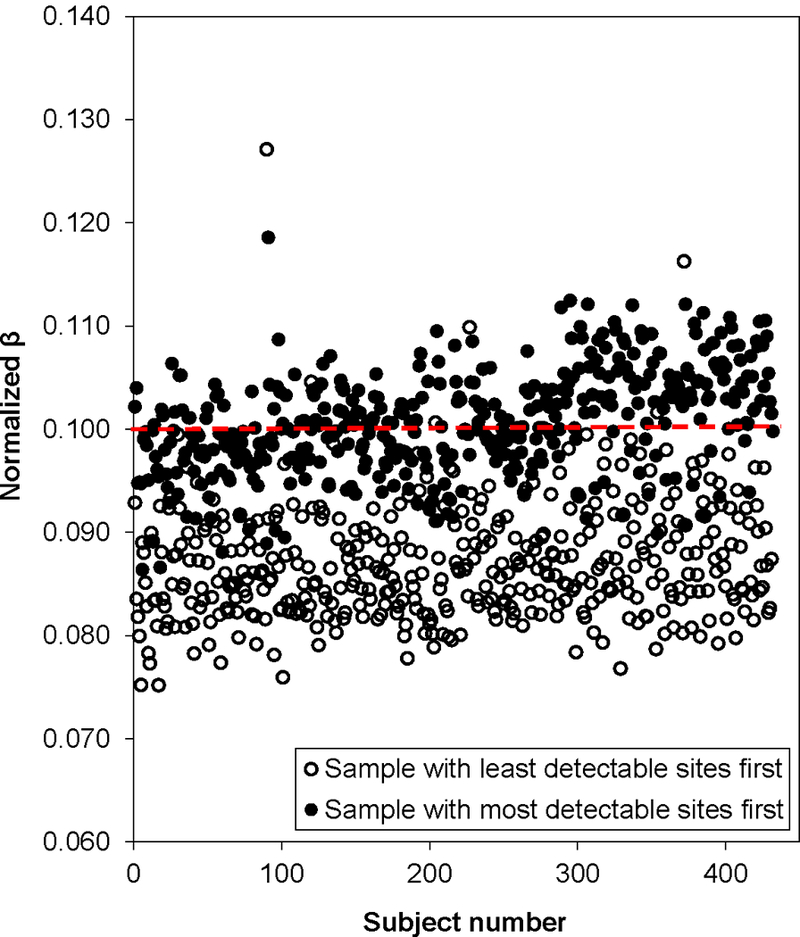
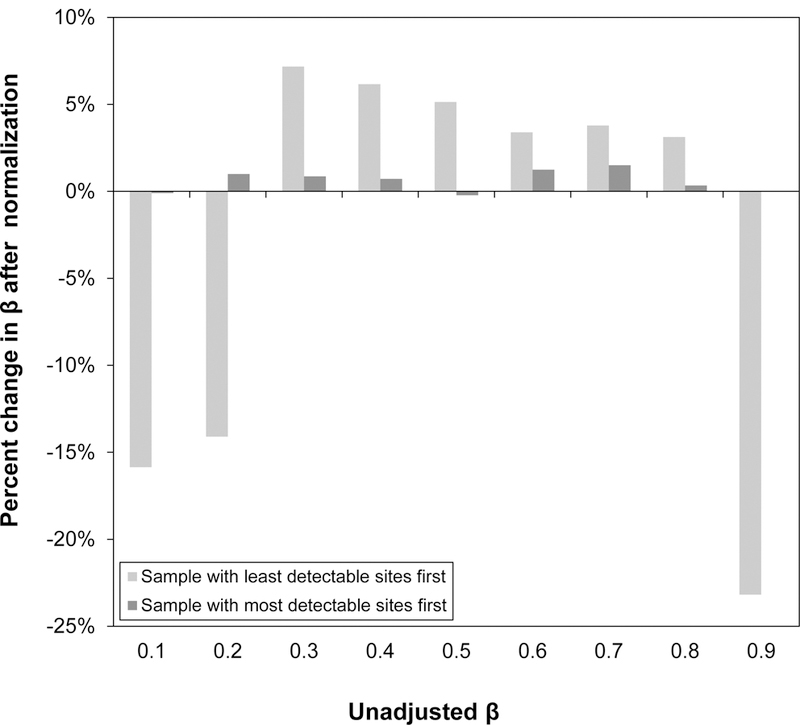
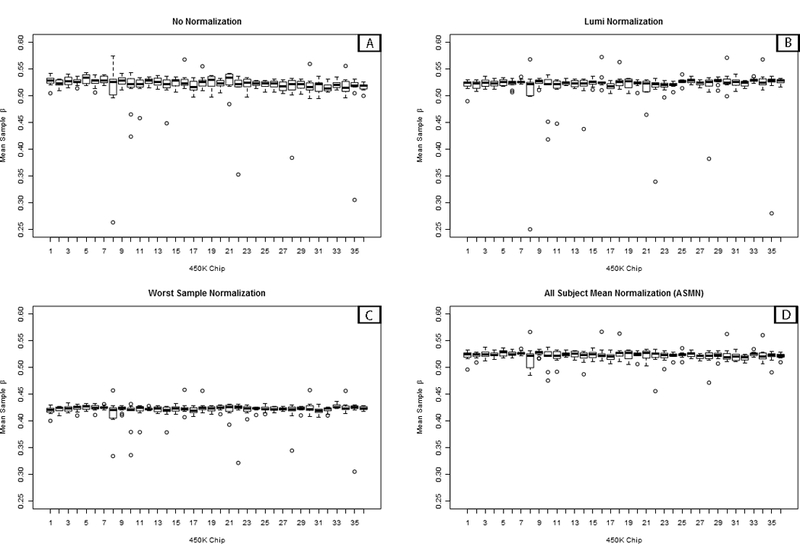
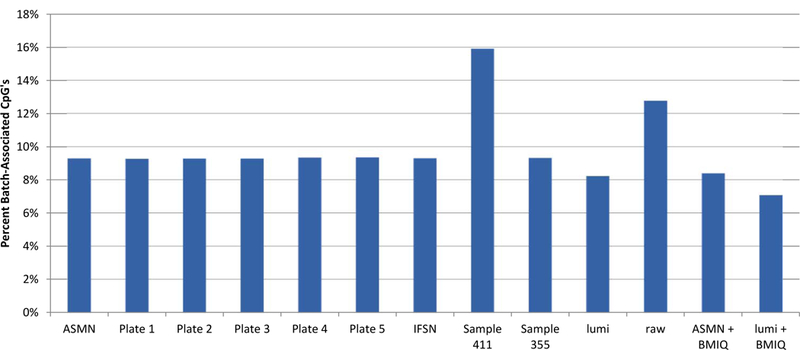
Analysis of epigenetic mechanisms, particularly DNA methylation, is of increasing interest for epidemiologic studies examining disease etiology and impacts of environmental exposures. The Infinium HumanMethylation450 BeadChip(®) (450K), which interrogates over 480,000 CpG sites and is relatively cost effective, has become a popular tool to characterize the DNA methylome. For large-scale studies, minimizing technical variability and potential bias is paramount. The goal of this paper was to evaluate the performance of several existing and novel color channel normalizations designed to reduce technical variability and batch effects in 450K analysis from a large population study. Comparative assessment of 10 normalization procedures included the GenomeStudio(®) Illumina procedure, the lumi smooth quantile approach, and the newly proposed All Sample Mean Normalization (ASMN). We also examined the performance of normalizations in combination with correction for the two types of Infinium chemistry utilized on the 450K array. We observed that the performance of the GenomeStudio(®) normalization procedure was highly variable and dependent on the quality of the first sample analyzed in an experiment, which is used as a reference in this procedure. While the lumi normalization was able to decrease batch variability, it increased variation among technical replicates, potentially reducing biologically meaningful findings. The proposed ASMN procedure performed consistently well, both at reducing batch effects and improving replicate comparability. In summary, the ASMN procedure can improve existing color channel normalization, especially for large epidemiologic studies, and can be successfully implemented to enhance a 450K DNA methylation data pipeline.
Keywords: ASMN; DNA methylome; bias correction; epigenetics; microarray; pipeline; technical variability.
Conflict of interest statement
The authors state that they have no potential conflicts of interest.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources