

Towards precision medicine: advances in computational approaches for the analysis of human variants
- PMID: 23962656
- PMCID: PMC3807015
- DOI: 10.1016/j.jmb.2013.08.008
Towards precision medicine: advances in computational approaches for the analysis of human variants
Abstract
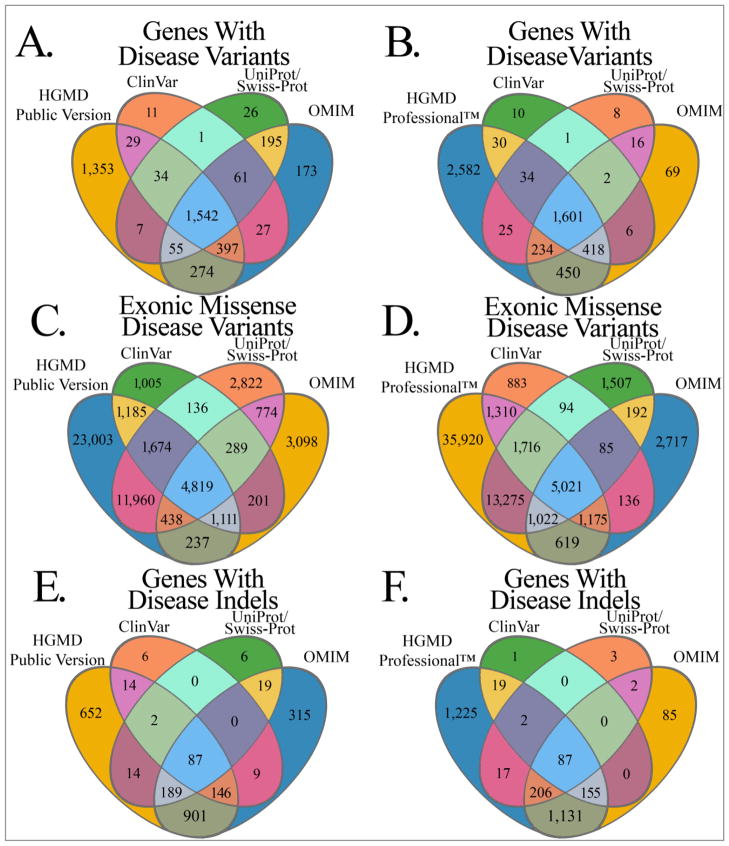
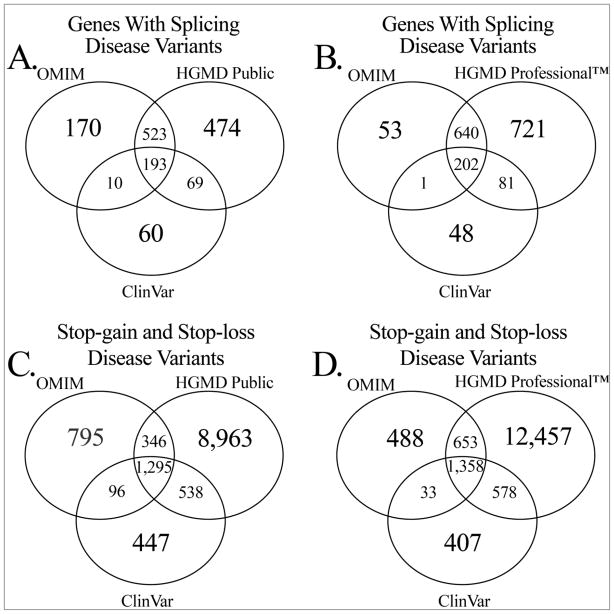
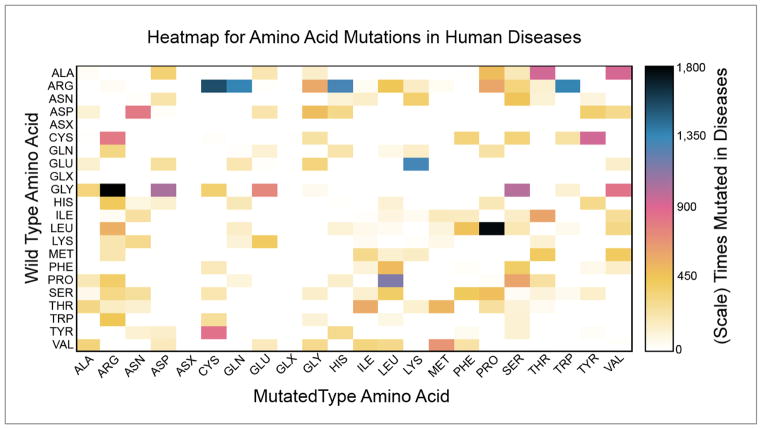
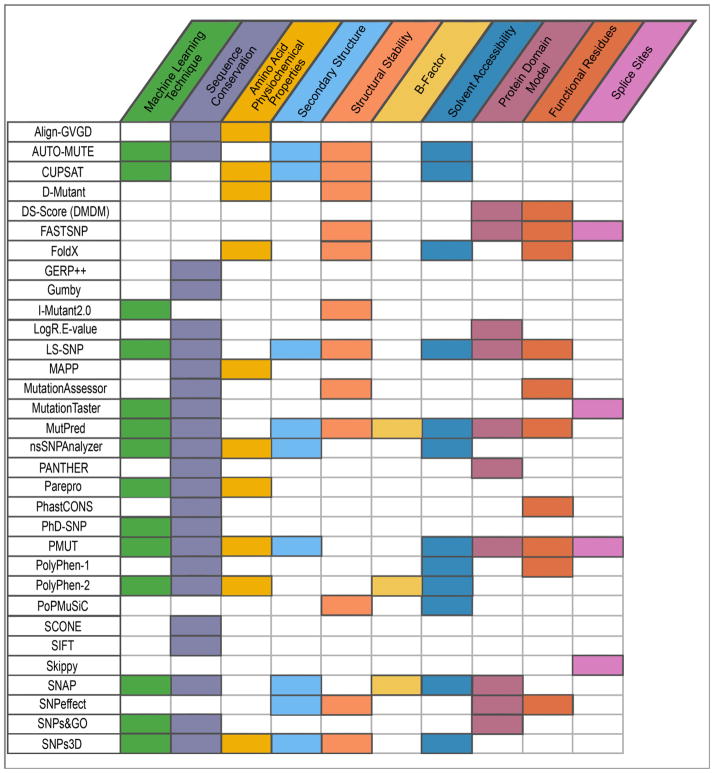
Variations and similarities in our individual genomes are part of our history, our heritage, and our identity. Some human genomic variants are associated with common traits such as hair and eye color, while others are associated with susceptibility to disease or response to drug treatment. Identifying the human variations producing clinically relevant phenotypic changes is critical for providing accurate and personalized diagnosis, prognosis, and treatment for diseases. Furthermore, a better understanding of the molecular underpinning of disease can lead to development of new drug targets for precision medicine. Several resources have been designed for collecting and storing human genomic variations in highly structured, easily accessible databases. Unfortunately, a vast amount of information about these genetic variants and their functional and phenotypic associations is currently buried in the literature, only accessible by manual curation or sophisticated text text-mining technology to extract the relevant information. In addition, the low cost of sequencing technologies coupled with increasing computational power has enabled the development of numerous computational methodologies to predict the pathogenicity of human variants. This review provides a detailed comparison of current human variant resources, including HGMD, OMIM, ClinVar, and UniProt/Swiss-Prot, followed by an overview of the computational methods and techniques used to leverage the available data to predict novel deleterious variants. We expect these resources and tools to become the foundation for understanding the molecular details of genomic variants leading to disease, which in turn will enable the promise of precision medicine.
Keywords: GWAS; Genome Wide Association Studies; HGVS; Human Genome Variation Society; LSDB; NCBI; National Center for Biotechnology Information; SVM; databases for human variants; function prediction; human disease variants; locus-specific database; support vector machine.
© 2013.
Figures
References
-
- Mirnezami R, Nicholson J, Darzi A. Preparing for precision medicine. N Engl J Med. 2012;366(6):489–91. - PubMed
-
- Yong E. We Gained Hope. The Story of Lilly Grossman’s Genome. 2013 [cited 2013 May 2nd]; Available from: http://phenomena.nationalgeographic.com/2013/03/11/we-gained-hope-the-st...
-
- A one-in-a million disease; a lifesaving bone marrow transplant - given twice. 2012 [cited 2013 May 23rd]; Available from: http://www.jsonline.com/features/health/a-gift-of-life--given-twice-6f4a....
-
- A genetic test solves a hereditary mystery and saves a life 2012. 2012 Dec 07; [Available from: http://www.theglobeandmail.com/news/national/time-to-lead/a-genetic-test...
-
- Collins FS, Morgan M, Patrinos A. The Human Genome Project: lessons from large-scale biology. Science. 2003;300(5617):286–90. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
