

Inflammatory process in Alzheimer's Disease
- PMID: 23964211
- PMCID: PMC3741576
- DOI: 10.3389/fnint.2013.00059
Inflammatory process in Alzheimer's Disease
Abstract
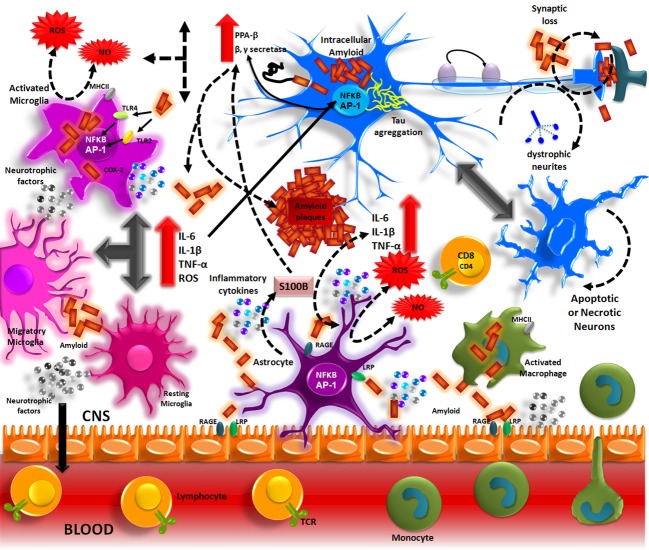
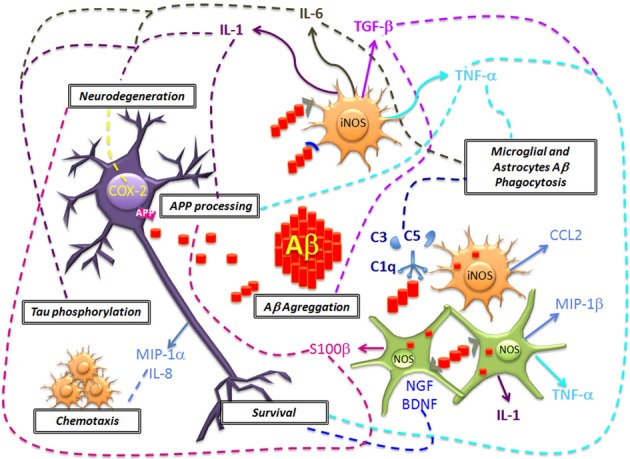
Alzheimer Disease (AD) is a neurodegenerative disorder and the most common form of dementia. Histopathologically is characterized by the presence of two major hallmarks, the intracellular neurofibrillary tangles (NFTs) and extracellular neuritic plaques (NPs) surrounded by activated astrocytes and microglia. NFTs consist of paired helical filaments of truncated tau protein that is abnormally hyperphosphorylated. The main component in the NP is the amyloid-β peptide (Aβ), a small fragment of 40-42 amino acids with a molecular weight of 4 kD. It has been proposed that the amyloid aggregates and microglia activation are able to favor the neurodegenerative process observed in AD patients. However, the role of inflammation in AD is controversial, because in early stages the inflammation could have a beneficial role in the pathology, since it has been thought that the microglia and astrocytes activated could be involved in Aβ clearance. Nevertheless the chronic activation of the microglia has been related with an increase of Aβ and possibly with tau phosphorylation. Studies in AD brains have shown an upregulation of complement molecules, pro-inflammatory cytokines, acute phase reactants and other inflammatory mediators that could contribute with the neurodegenerative process. Clinical trials and animal models with non-steroidal anti-inflammatory drugs (NSAIDs) indicate that these drugs may decrease the risk of developing AD and apparently reduce Aβ deposition. Finally, further studies are needed to determine whether treatment with anti-inflammatory strategies, may decrease the neurodegenerative process that affects these patients.
Keywords: Alzheimer disease; amyloid-β; anti-inflammatory strategies; astrocyte; microglia; neurodegeneration; neuroinflammation; pro-inflammatory cytokine.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous