

Recent advances in obesity-induced inflammation and insulin resistance
- PMID: 23964268
- PMCID: PMC3737462
- DOI: 10.3389/fendo.2013.00093
Recent advances in obesity-induced inflammation and insulin resistance
Abstract
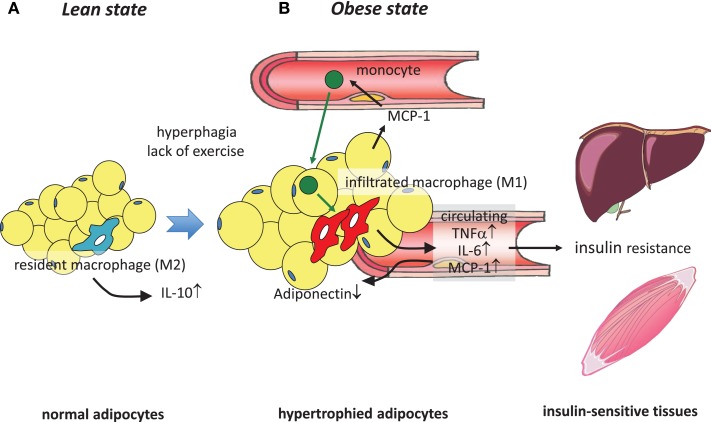
It has been demonstrated in rodents and humans that chronic inflammation characterized by macrophage infiltration occurs mainly in adipose tissue or liver during obesity, in which activation of immune cells is closely associated with insulin sensitivity. Macrophages can be classified as classically activated (M1) macrophages that support microbicidal activity or alternatively activated (M2) macrophages that support allergic and antiparasitic responses. In the context of insulin action, M2 macrophages sustain insulin sensitivity by secreting IL-4 and IL-10, while M1 macrophages induce insulin resistance through the secretion of proinflammatory cytokines, such as TNFα. Polarization of M1/M2 is controlled by various dynamic functions of other immune cells. It has been demonstrated that, in a lean state, TH2 cells, Treg cells, natural killer T cells, or eosinophils contribute to the M2 activation of macrophages by secreting IL-4 or IL-10. In contrast, obesity causes alteration of the constituent immune cells, in which TH1 cells, B cells, neutrophils, or mast cells induce M1 activation of macrophages by the elevated secretion of TNFα and IFNγ. Increased secretion of TNFα and free fatty acids from hypertrophied adipocytes also contributes to the M1 activation of macrophages. Since obesity-induced insulin resistance is established by macrophage infiltration and the activation of immune cells inside tissues, identification of the factors that regulate accumulation and the intracellular signaling cascades that define polarization of M1/M2 would be indispensable. Regulation of these factors would lead to the pharmacological inhibition of obesity-induced insulin resistance. In this review, we introduce molecular mechanisms relevant to the pathophysiology and review the most recent studies of clinical applications targeting chronic inflammation.
Keywords: TNFα; adipose tissue; chronic inflammation; insulin resistance; macrophages; obesity.
Figures


References
-
- Ebstein W. Zur therapie des diabetes mellitus, insbesondere uber die anwendeng der salicylauren natron bei demselben. Klin Wochenschr (1876) 13:337–40
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources