

Developing therapeutic approaches for metachromatic leukodystrophy
- PMID: 23966770
- PMCID: PMC3743609
- DOI: 10.2147/DDDT.S15467
Developing therapeutic approaches for metachromatic leukodystrophy
Abstract
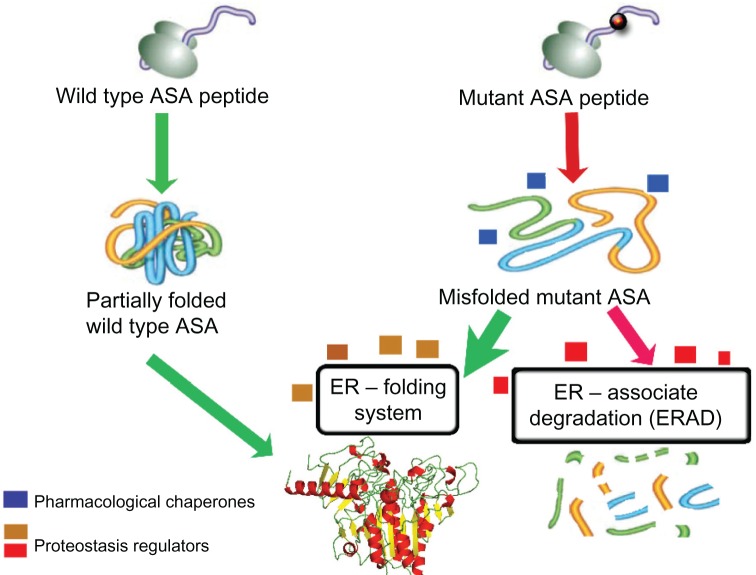
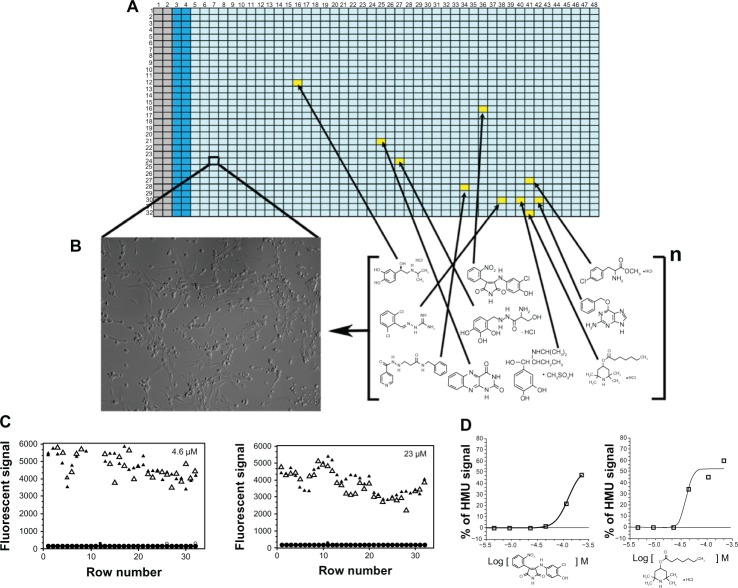
Metachromatic leukodystrophy (MLD) is an autosomal recessive lysosomal disorder caused by the deficiency of arylsulfatase A (ASA), resulting in impaired degradation of sulfatide, an essential sphingolipid of myelin. The clinical manifestations of MLD are characterized by progressive demyelination and subsequent neurological symptoms resulting in severe debilitation. The availability of therapeutic options for treating MLD is limited but expanding with a number of early stage clinical trials already in progress. In the development of therapeutic approaches for MLD, scientists have been facing a number of challenges including blood-brain barrier (BBB) penetration, safety issues concerning therapies targeting the central nervous system, uncertainty regarding the ideal timing for intervention in the disease course, and the lack of more in-depth understanding of the molecular pathogenesis of MLD. Here, we discuss the current status of the different approaches to developing therapies for MLD. Hematopoietic stem cell transplantation has been used to treat MLD patients, utilizing both umbilical cord blood and bone marrow sources. Intrathecal enzyme replacement therapy and gene therapies, administered locally into the brain or by generating genetically modified hematopoietic stem cells, are emerging as novel strategies. In pre-clinical studies, different cell delivery systems including microencapsulated cells or selectively neural cells have shown encouraging results. Small molecules that are more likely to cross the BBB can be used as enzyme enhancers of diverse ASA mutants, either as pharmacological chaperones, or proteostasis regulators. Specific small molecules may also be used to reduce the biosynthesis of sulfatides, or target different affected downstream pathways secondary to the primary ASA deficiency. Given the progressive neurodegenerative aspects of MLD, also seen in other lysosomal diseases, current and future therapeutic strategies will be complementary, whether used in combination or separately at specific stages of the disease course, to produce better outcomes for patients afflicted with this devastating inherited disorder.
Keywords: arylsulfatase A; enzyme enhancement therapy; enzyme replacement therapy; gene therapy; metachromatic leukodystrophy; small molecules.
Figures

References
-
- Austin J, McAfee D, Armstrong D, O’Rourke M, Shearer L, Bachhawat B. Low sulfatase activities in metachromatic leukodystrophy (MLD). A controlled study of enzymes in 9 living and 4 autopsied patients with MLD. Trans Am Neurol Assoc. 1964;89:147–150. - PubMed
-
- Aggarwal S, Yurlova L, Simons M. Central nervous system myelin: structure, synthesis and assembly. Trends Cell Biol. 2011;21(10):585–593. - PubMed
-
- Faust PL, Kaye EM, Powers JM. Myelin lesions associated with lysosomal and peroxisomal disorders. Expert Rev Neurother. 2010;10(9):1449–1466. - PubMed
-
- Gieselmann V. Metachromatic leukodystrophy: genetics, pathogenesis and therapeutic options. Acta Paediatr Suppl. 2008;97(457):15–21. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
