

The role of selection in shaping diversity of natural M. tuberculosis populations
- PMID: 23966858
- PMCID: PMC3744410
- DOI: 10.1371/journal.ppat.1003543
The role of selection in shaping diversity of natural M. tuberculosis populations
Erratum in
- PLoS Pathog. 2013 Aug;9(8). doi:10.1371/annotation/cff22061-44d5-4301-b853-41702d160203. Holmes, Eddie C [corrected to Holmes, Edward C]
Abstract
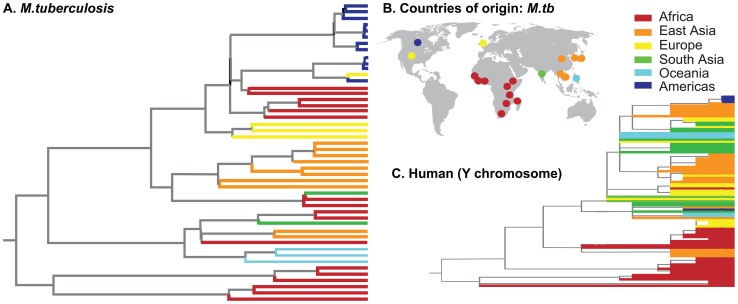
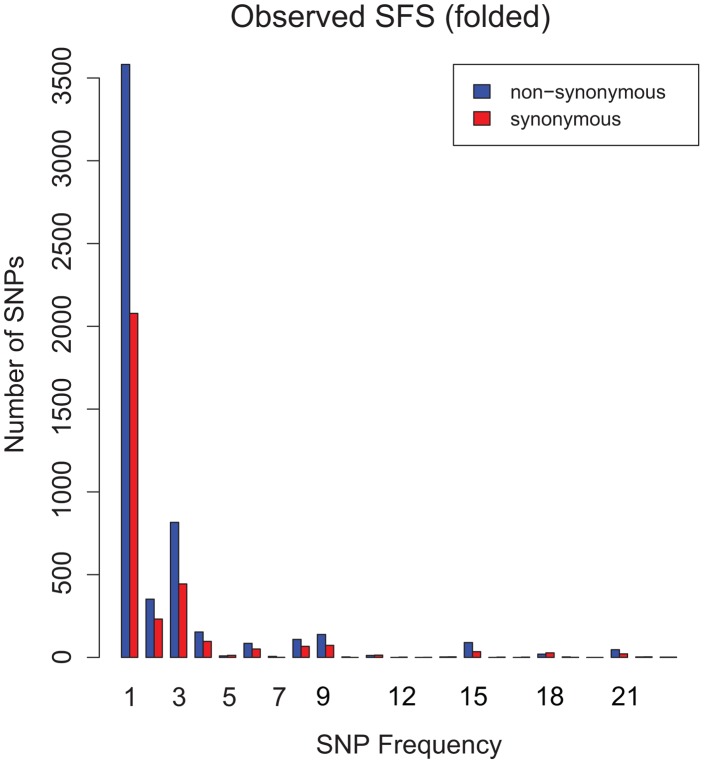
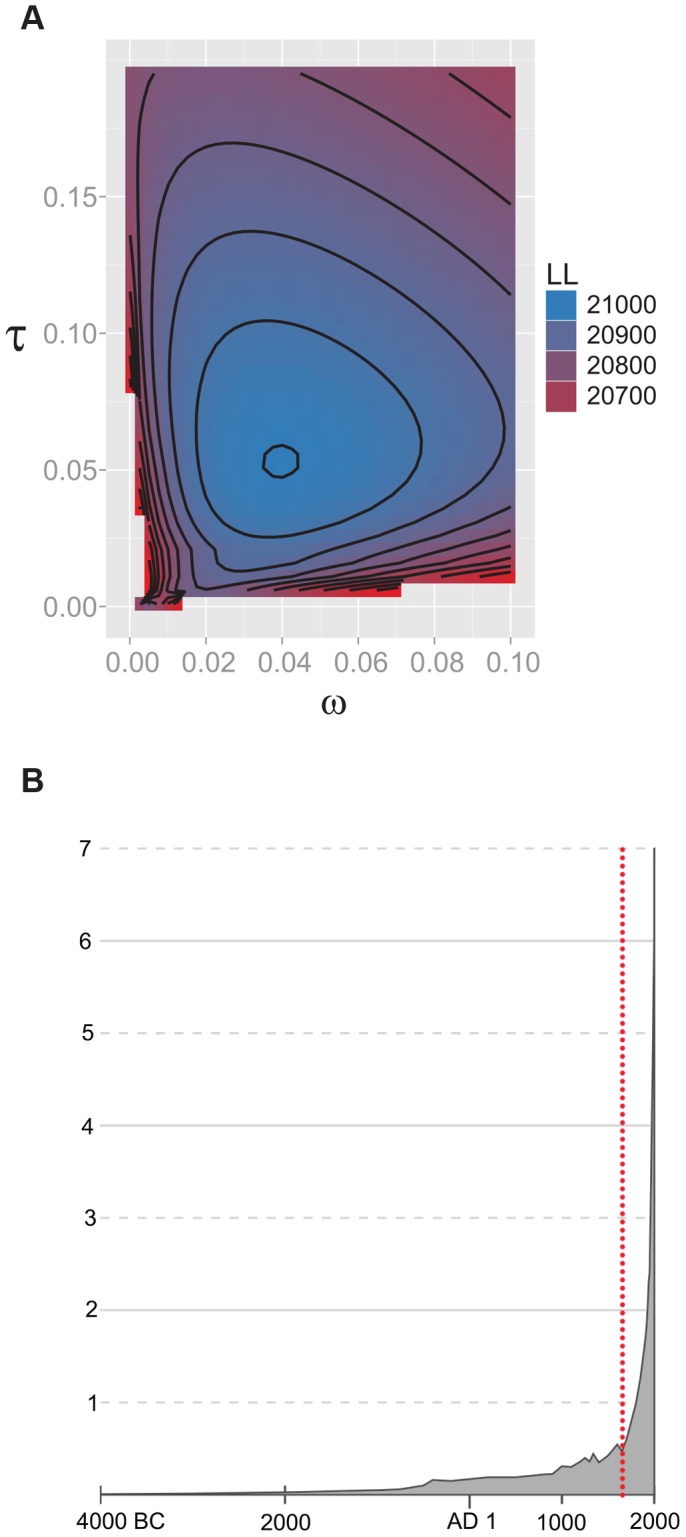
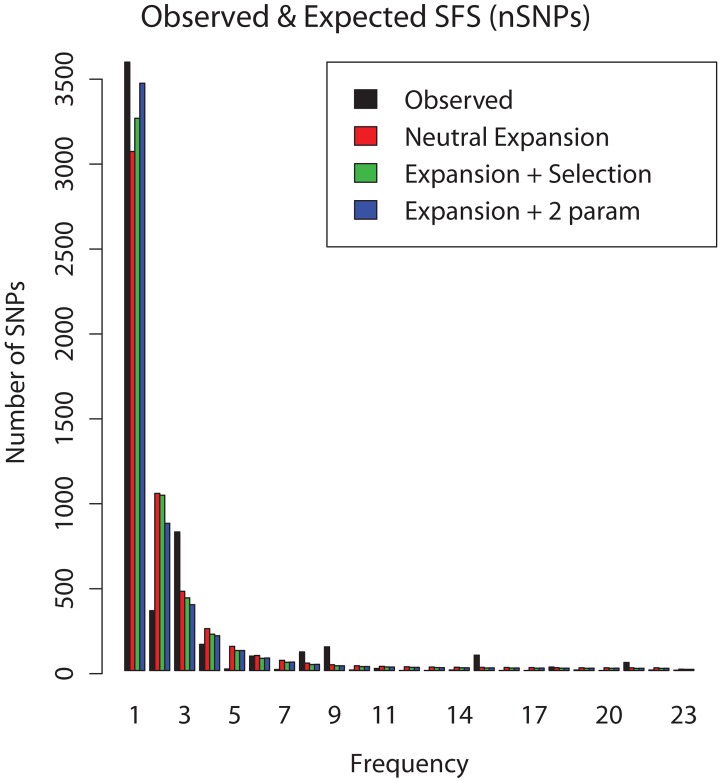
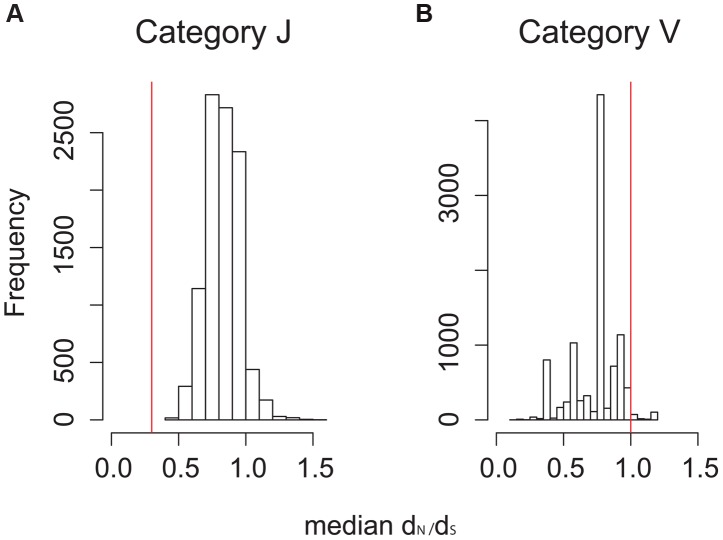
Mycobacterium tuberculosis (M.tb), the cause of tuberculosis (TB), is estimated to infect a new host every second. While analyses of genetic data from natural populations of M.tb have emphasized the role of genetic drift in shaping patterns of diversity, the influence of natural selection on this successful pathogen is less well understood. We investigated the effects of natural selection on patterns of diversity in 63 globally extant genomes of M.tb and related pathogenic mycobacteria. We found evidence of strong purifying selection, with an estimated genome-wide selection coefficient equal to -9.5 × 10(-4) (95% CI -1.1 × 10(-3) to -6.8 × 10(-4)); this is several orders of magnitude higher than recent estimates for eukaryotic and prokaryotic organisms. We also identified different patterns of variation across categories of gene function. Genes involved in transport and metabolism of inorganic ions exhibited very low levels of non-synonymous polymorphism, equivalent to categories under strong purifying selection (essential and translation-associated genes). The highest levels of non-synonymous variation were seen in a group of transporter genes, likely due to either diversifying selection or local selective sweeps. In addition to selection, we identified other important influences on M.tb genetic diversity, such as a 25-fold expansion of global M.tb populations coincident with explosive growth in human populations (estimated timing 1684 C.E., 95% CI 1620-1713 C.E.). These results emphasize the parallel demographic histories of this obligate pathogen and its human host, and suggest that the dominant effect of selection on M.tb is removal of novel variants, with exceptions in an interesting group of genes involved in transportation and defense. We speculate that the hostile environment within a host imposes strict demands on M.tb physiology, and thus a substantial fitness cost for most new mutations. In this respect, obligate bacterial pathogens may differ from other host-associated microbes such as symbionts.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
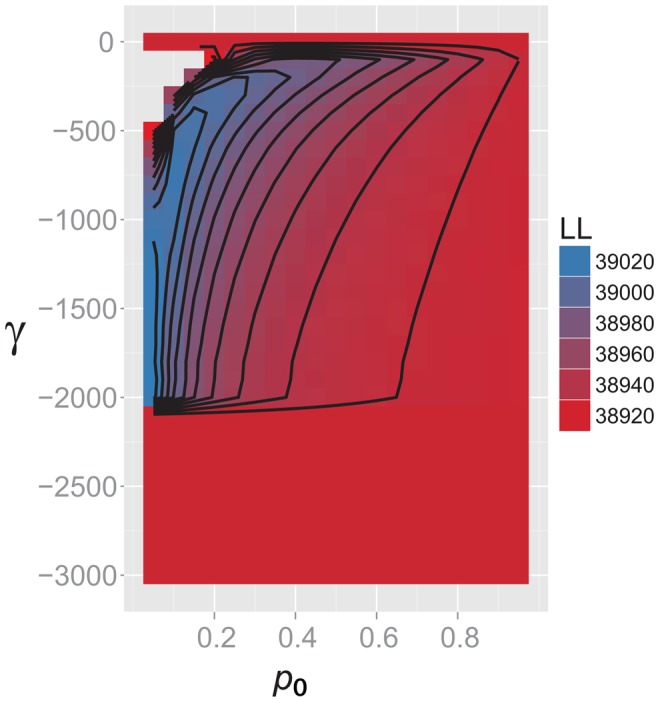
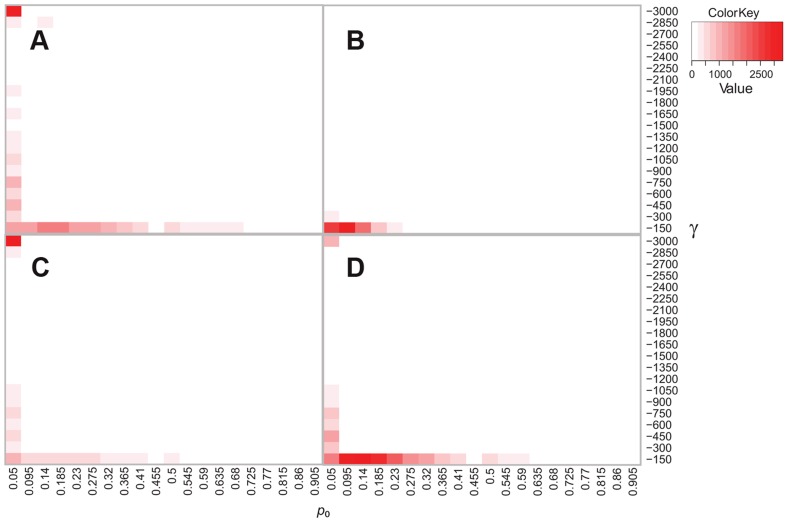
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
