

Detecting association of rare variants by testing an optimally weighted combination of variants for quantitative traits in general families
- PMID: 23968488
- PMCID: PMC3932153
- DOI: 10.1111/ahg.12038
Detecting association of rare variants by testing an optimally weighted combination of variants for quantitative traits in general families
Abstract
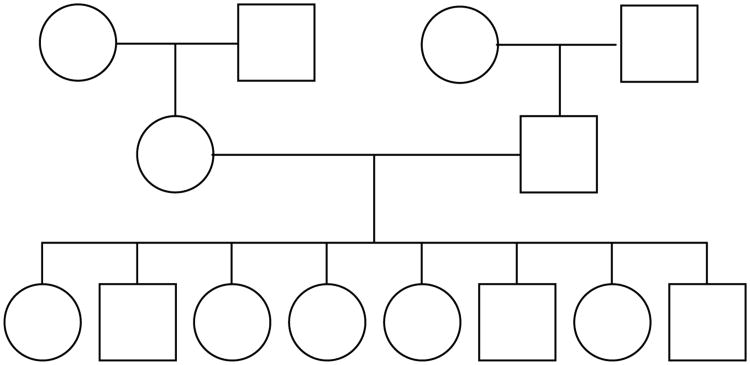
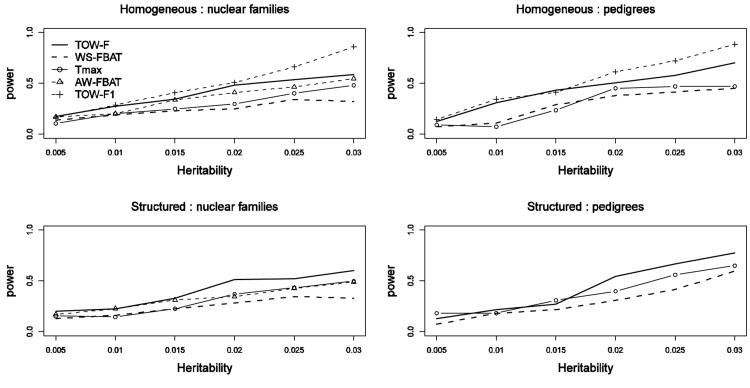
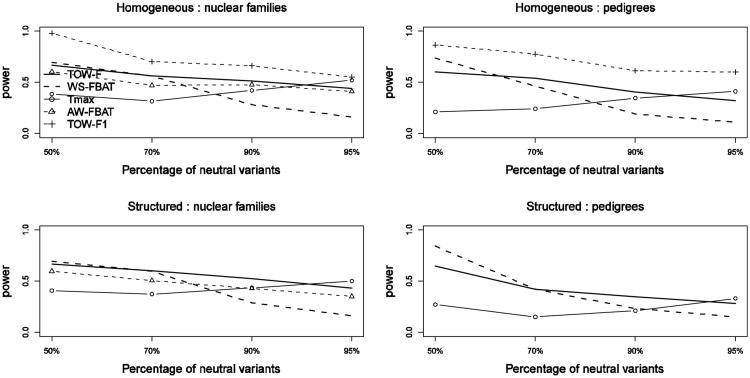
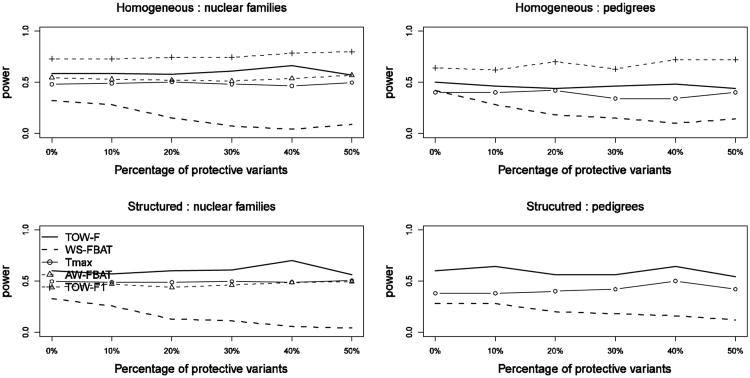
Although next-generation sequencing technology allows sequencing the whole genome of large groups of individuals, the development of powerful statistical methods for rare variant association studies is still underway. Even though many statistical methods have been developed for mapping rare variants, most of these methods are for unrelated individuals only, whereas family data have been shown to improve power to detect rare variants. The majority of the existing methods for unrelated individuals is essentially testing the effect of a weighted combination of variants with different weighting schemes. The performance of these methods depends on the weights being used. Recently, researchers proposed a test for Testing the effect of an Optimally Weighted combination of variants (TOW) for unrelated individuals. In this article, we extend our previously developed TOW for unrelated individuals to family-based data and propose a novel test for Testing the effect of an Optimally Weighted combination of variants for Family-based designs (TOW-F). The optimal weights are analytically derived. The results of extensive simulation studies show that TOW-F is robust to population stratification in a wide range of population structures, is robust to the direction and magnitude of the effects of causal variants, and is relatively robust to the percentage of neutral variants.
Keywords: Rare variants; association studies; general families; population stratification; quantitative traits.
© 2013 John Wiley & Sons Ltd/University College London.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
