

BMP9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia
- PMID: 23972370
- PMCID: PMC3769931
- DOI: 10.1016/j.ajhg.2013.07.004
BMP9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia
Abstract
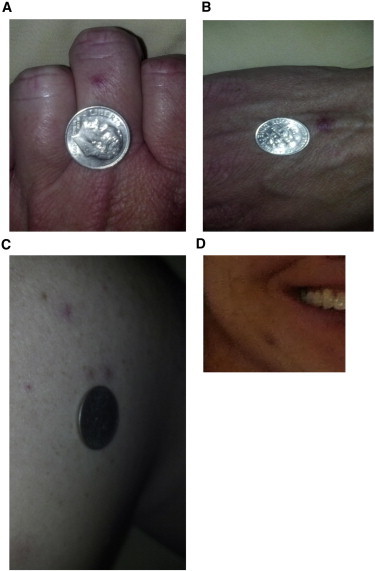
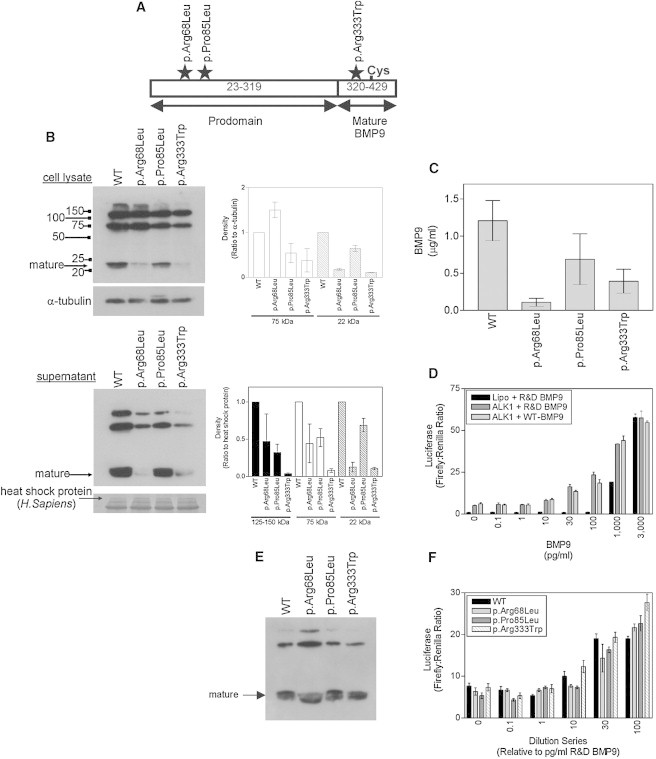
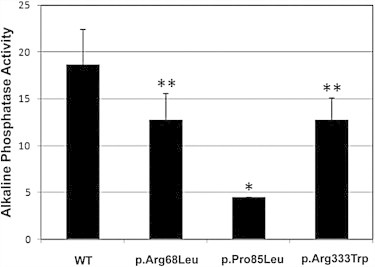
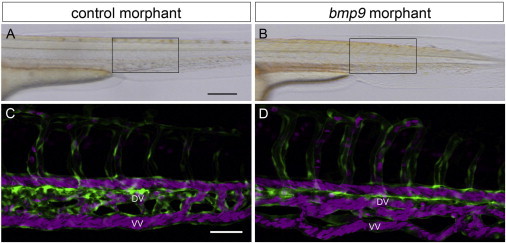
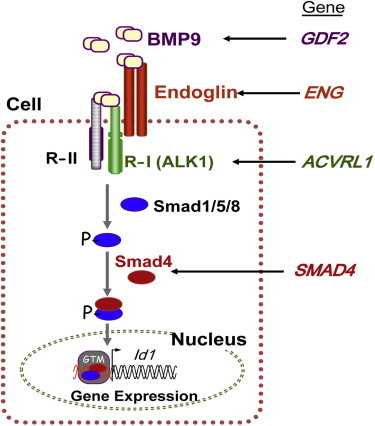
Hereditary hemorrhagic telangiectasia (HHT), the most common inherited vascular disorder, is caused by mutations in genes involved in the transforming growth factor beta (TGF-β) signaling pathway (ENG, ACVRL1, and SMAD4). Yet, approximately 15% of individuals with clinical features of HHT do not have mutations in these genes, suggesting that there are undiscovered mutations in other genes for HHT and possibly vascular disorders with overlapping phenotypes. The genetic etiology for 191 unrelated individuals clinically suspected to have HHT was investigated with the use of exome and Sanger sequencing; these individuals had no mutations in ENG, ACVRL1, and SMAD4. Mutations in BMP9 (also known as GDF2) were identified in three unrelated probands. These three individuals had epistaxis and dermal lesions that were described as telangiectases but whose location and appearance resembled lesions described in some individuals with RASA1-related disorders (capillary malformation-arteriovenous malformation syndrome). Analyses of the variant proteins suggested that mutations negatively affect protein processing and/or function, and a bmp9-deficient zebrafish model demonstrated that BMP9 is involved in angiogenesis. These data confirm a genetic cause of a vascular-anomaly syndrome that has phenotypic overlap with HHT.
Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Richards-Yutz J., Grant K., Chao E.C., Walther S.E., Ganguly A. Update on molecular diagnosis of hereditary hemorrhagic telangiectasia. Hum. Genet. 2010;128:61–77. - PubMed
-
- Shovlin C.L., Guttmacher A.E., Buscarini E., Faughnan M.E., Hyland R.H., Westermann C.J., Kjeldsen A.D., Plauchu H. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome) Am. J. Med. Genet. 2000;91:66–67. - PubMed
-
- McDonald M.T., Papenberg K.A., Ghosh S., Glatfelter A.A., Biesecker B.B., Helmbold E.A., Markel D.S., Zolotor A., McKinnon W.C., Vanderstoep J.L. A disease locus for hereditary haemorrhagic telangiectasia maps to chromosome 9q33-34. Nat. Genet. 1994;6:197–204. - PubMed
-
- Johnson D.W., Berg J.N., Gallione C.J., McAllister K.A., Warner J.P., Helmbold E.A., Markel D.S., Jackson C.E., Porteous M.E., Marchuk D.A. A second locus for hereditary hemorrhagic telangiectasia maps to chromosome 12. Genome Res. 1995;5:21–28. - PubMed
-
- Gallione C.J., Repetto G.M., Legius E., Rustgi A.K., Schelley S.L., Tejpar S., Mitchell G., Drouin E., Westermann C.J., Marchuk D.A. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4) Lancet. 2004;363:852–859. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
