

MC1R is a potent regulator of PTEN after UV exposure in melanocytes
- PMID: 23973372
- PMCID: PMC3792490
- DOI: 10.1016/j.molcel.2013.08.010
MC1R is a potent regulator of PTEN after UV exposure in melanocytes
Abstract
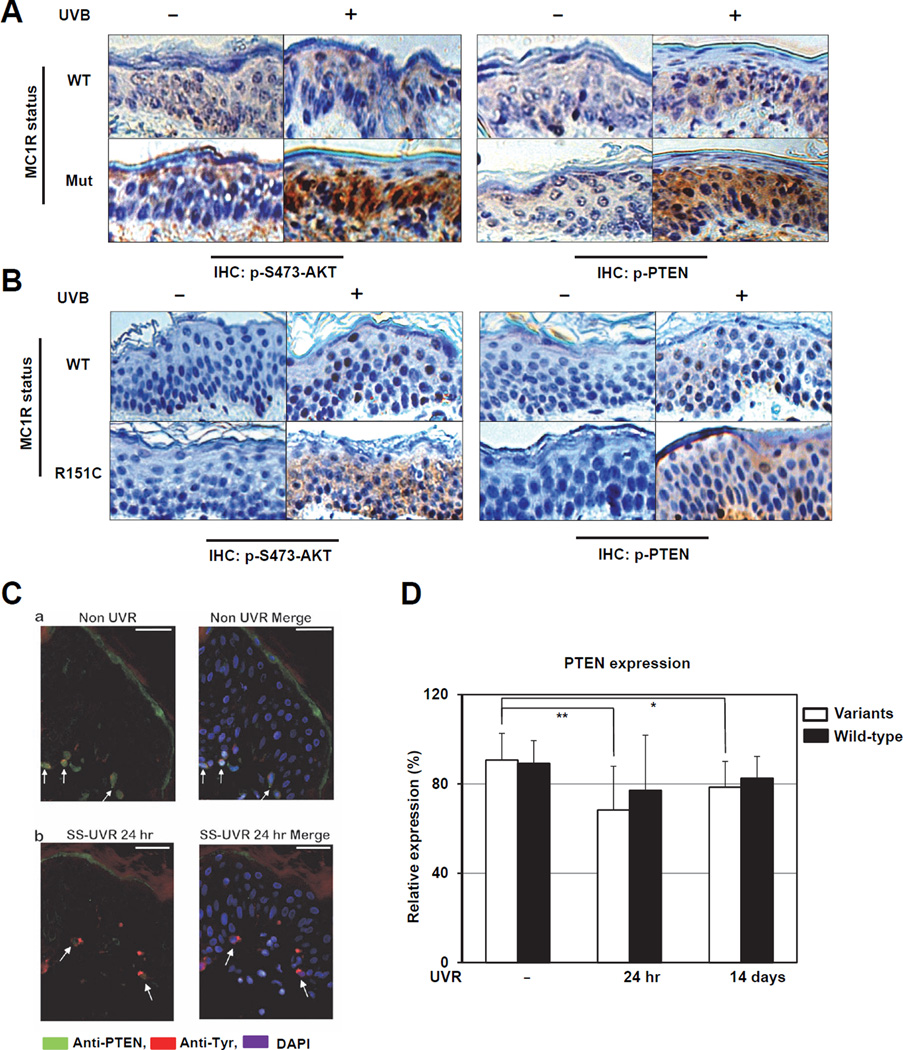
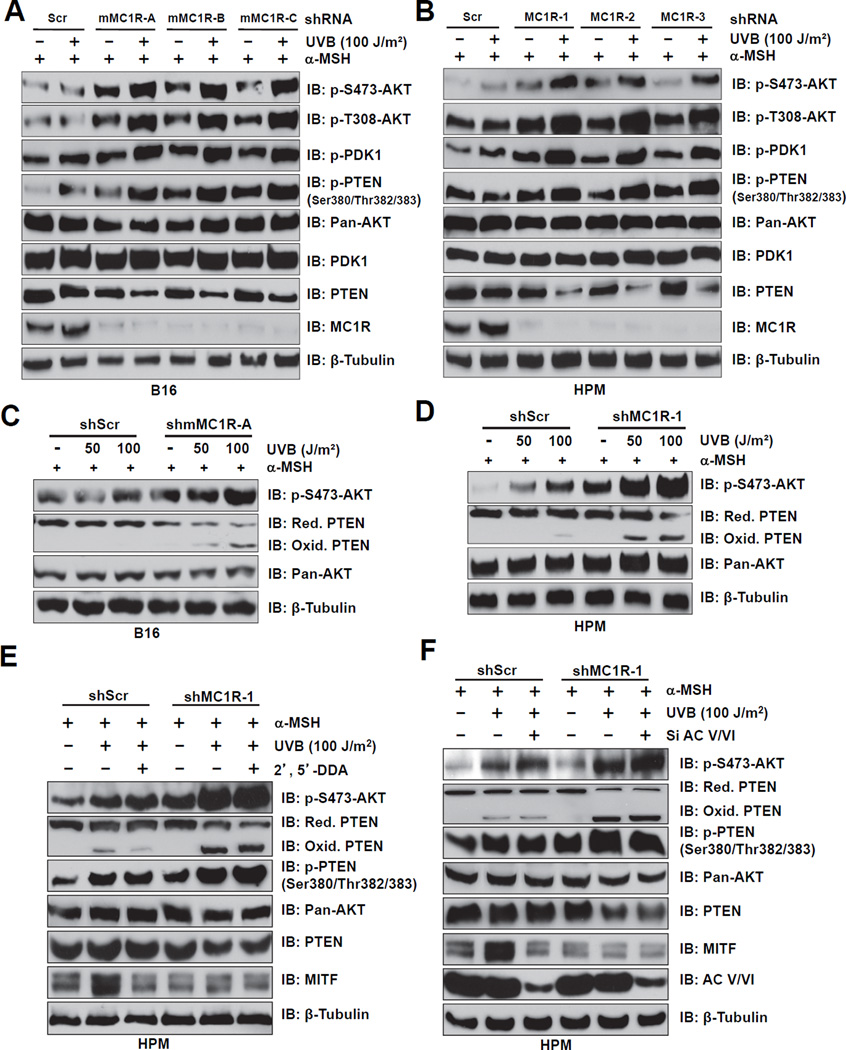
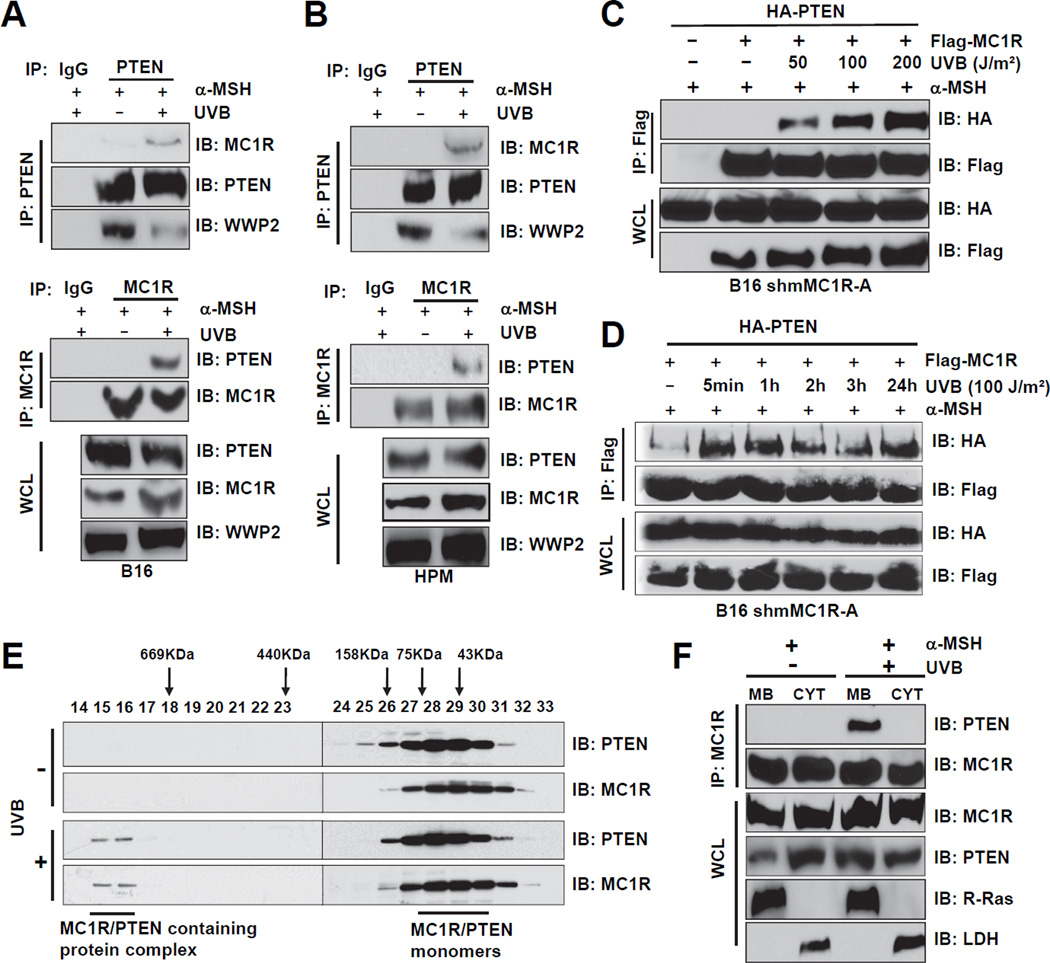
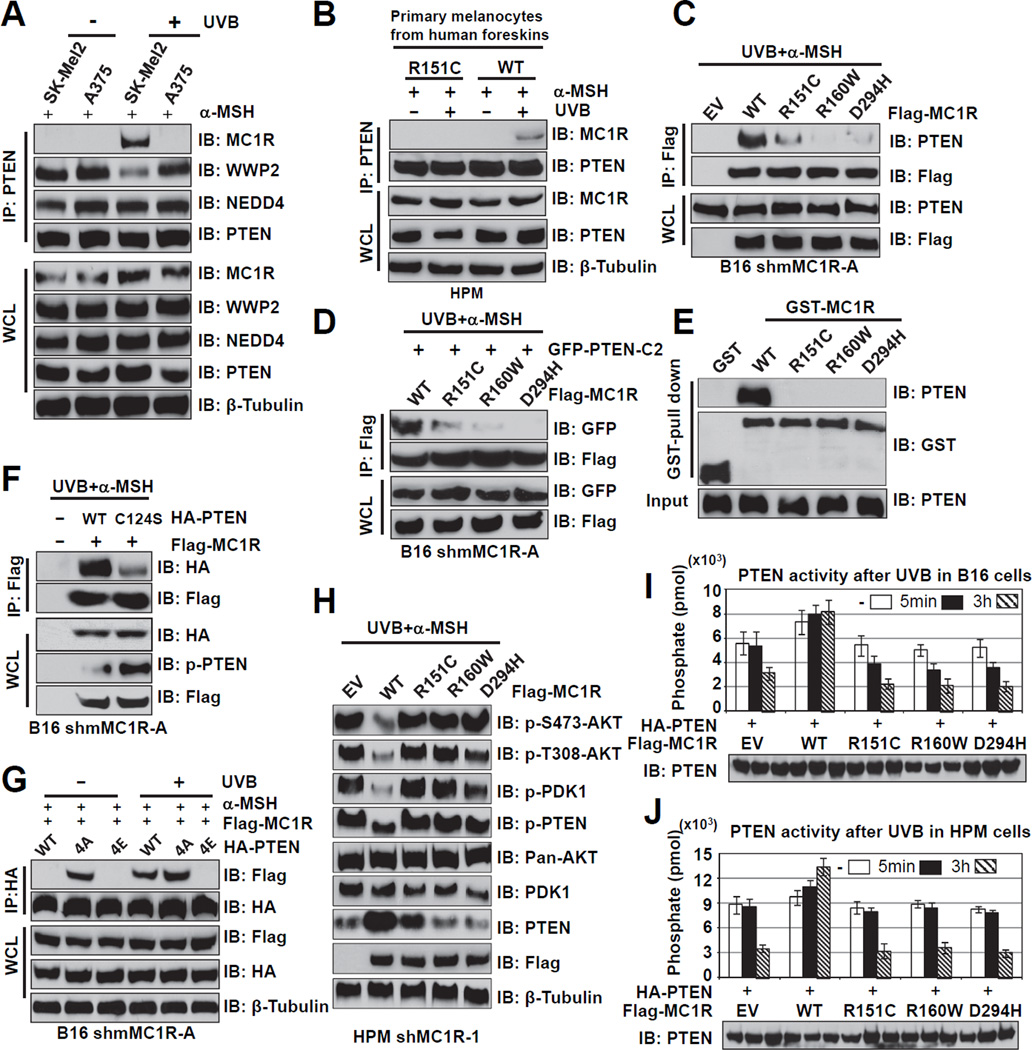
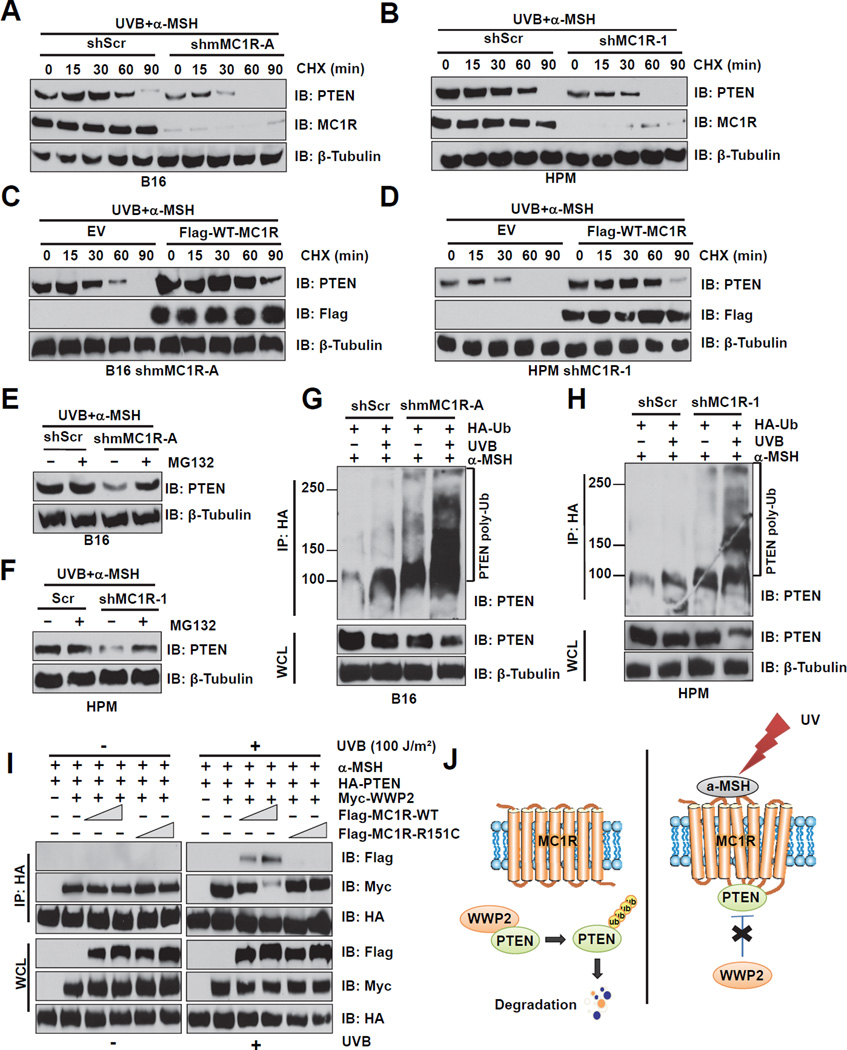
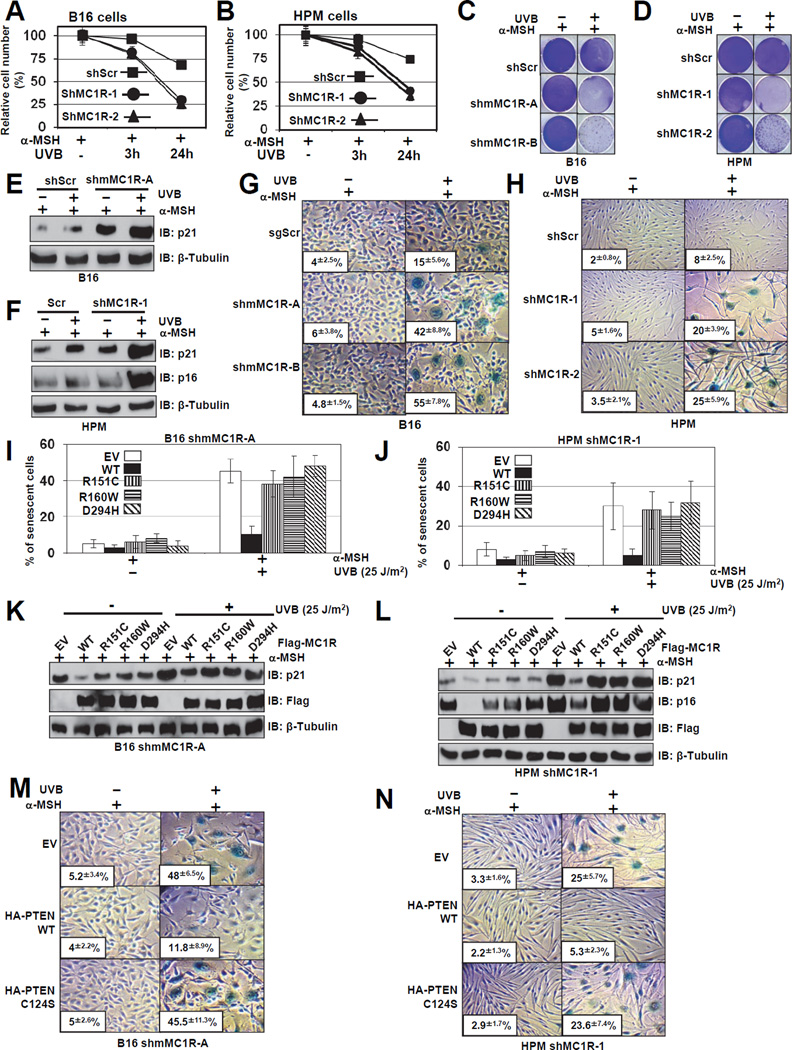
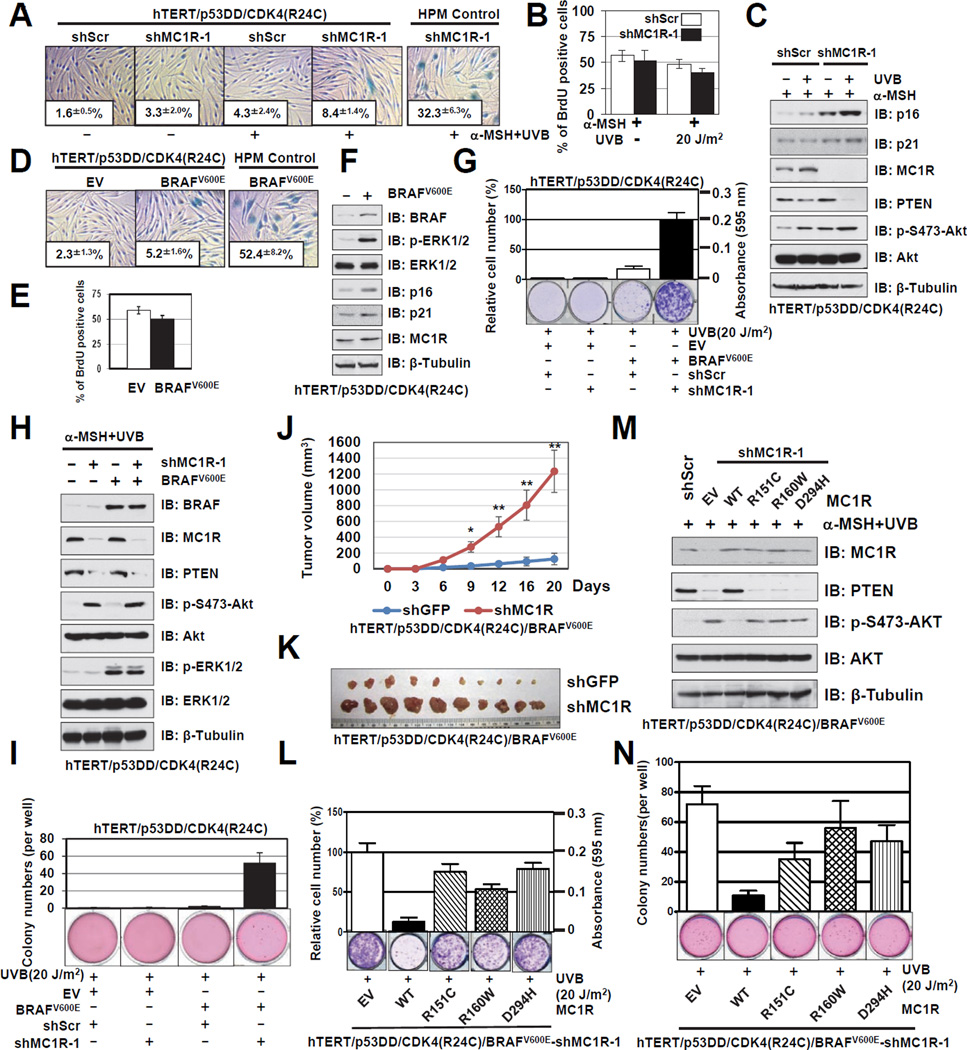
The individuals carrying melanocortin-1 receptor (MC1R) variants, especially those associated with red hair color, fair skin, and poor tanning ability (RHC trait), are more prone to melanoma; however, the underlying mechanism is poorly defined. Here, we report that UVB exposure triggers phosphatase and tensin homolog (PTEN) interaction with wild-type (WT), but not RHC-associated MC1R variants, which protects PTEN from WWP2-mediated degradation, leading to AKT inactivation. Strikingly, the biological consequences of the failure of MC1R variants to suppress PI3K/AKT signaling are highly context dependent. In primary melanocytes, hyperactivation of PI3K/AKT signaling leads to premature senescence; in the presence of BRAF(V600E), MC1R deficiency-induced elevated PI3K/AKT signaling drives oncogenic transformation. These studies establish the MC1R-PTEN axis as a central regulator for melanocytes' response to UVB exposure and reveal the molecular basis underlying the association between MC1R variants and melanomagenesis.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
References
-
- Ben-Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 2005;37:961–976. - PubMed
-
- Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, Stein H, Dorken B, Jenuwein T, Schmitt CA. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–665. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
